RESUMO
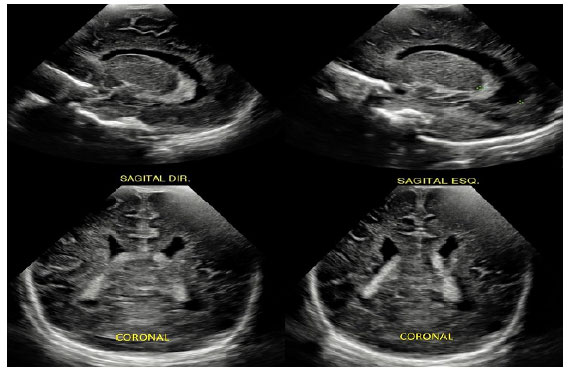
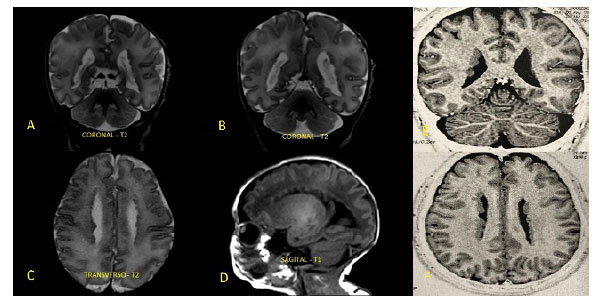
A heterotopia nodular periventricular (HP) é resultado da anormalidade na migração neuronal durante a gênese do sistema nervoso central, especialmente no período fetal. Apresenta-se assintomática no período neonatal, porém caracteriza-se, muitas vezes, como foco epileptiforme de difícil controle no decorrer da vida, causando impacto no desenvolvimento neuropsicomotor e na qualidade de vida da criança e de seu cuidador. Pelo fato de estar relacionada à anormalidade genética, muitas vezes derivada de padrão hereditário, pode-se predizer que pais acometidos poderão gerar prole com tal condição clínica. Nesse contexto, entretanto, o uso da ultrassonografia transfontanelar (USTF) no período neonatal como rastreio de HP em pacientes que possuem história familiar positiva ainda não é consenso na literatura atual. Assim, buscamos relatar o caso de HP diagnosticada ao acaso, em neonato assintomático, mas com genitora epiléptica portadora da mesma condição, não conhecida durante o pré-natal. A criança apresentou sinais sugestivos de ectopia neuronal em USTF solicitada devido a risco de hemorragia periventricular, sendo, posteriormente, confirmada com ressonância magnética de encéfalo.
Palavras-chave: Heterotopia nodular periventricular, Malformações do desenvolvimento cortical, Malformações do desenvolvimento cortical do grupo II, Ultrassonografia