RESUMO
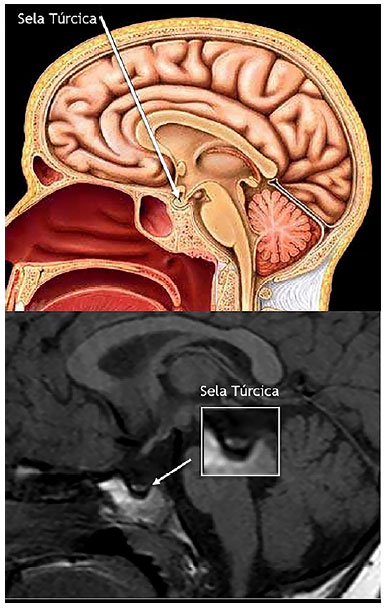
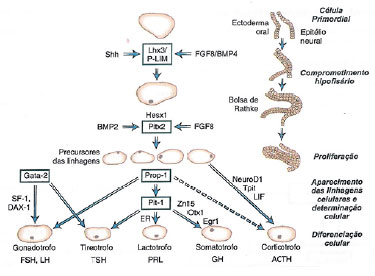
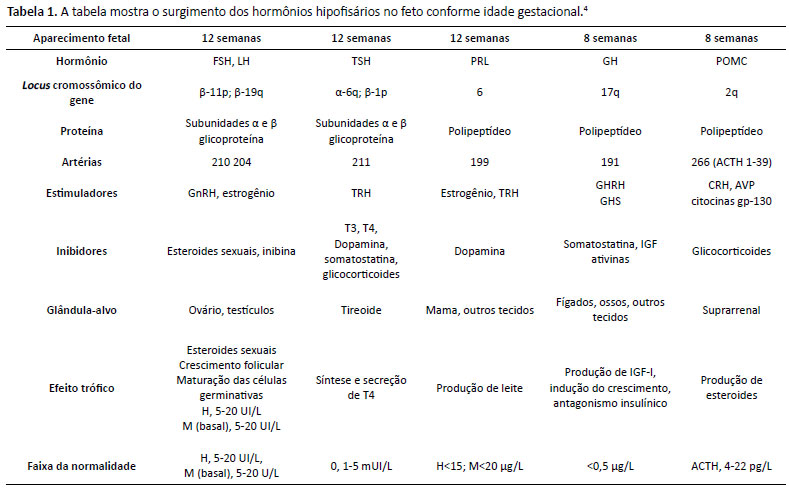
A deficiência na produção ou na ação de qualquer um dos hormônios da adeno-hipófise é denominada hipopituitarismo. Trata-se de diagnóstico desafiador e seus portadores apresentam maior mortalidade quando comparados à população geral, principalmente quando não recebem reposição hormonal adequada. Por meio da análise de prontuário de paciente do sexo feminino, habitante do Distrito Federal, 2 anos e 6 meses de vida, peso: 9,05kg (z-escore: -2,97); estatura: 80cm (z-escore: -3,07); IMC: 14,14 (z-escore: -1,27), foi encontrado quadro clínico de déficit pôndero-estatural e atraso do desenvolvimento neuropsicomotor, que, associado a exames laboratoriais e de imagem, levou ao diagnóstico de hipopituitarismo. Cabe ao médico, em especial o pediatra, estar atento para um diagnóstico tão singular.
Palavras-chave: Hipopituitarismo, Endocrinologia, Pediatria.