Relato de Caso
-
Ano 2023 -
Volume 13 -
Número
3
Deficiência de Lipase Ácida Lisossômica (LAL): análise enzimática em papel-filtro como ferramenta diagnóstica em paciente com diagnóstico prévio de doença de Niemann-Pick tipo C
Lysosomal Acid Lipase ( LAL) Deficiency: Enzyme Assay on Dried Blood Spot as Diagnostic Tool in a Patient Misdiagnosed as Niemann-Pick type C Disease
Marcella Borges1; Luissa Hikari Hayashi Araujo1; Bruno Lima1; Laura Vagnini2; Alberto Salles1; Guerino Pelicer Neto-Magalhães1; João Paulo Cristofolo1; João Paulo Freitas1; Pedro Ivo Aranas1; Jacqueline Harouche Rodrigues Fonseca3; Fernanda Seabra Souza Timm4; Charles Marques Lourenço1,5
RESUMO
INTRODUÇÃO: A deficiência de lipase acida lisossômica (LAL-D) é uma doença de armazenamento lisossômico envolvida no metabolismo do éster de colesterol. É uma causa genética pouco conhecida de cirrose, dislipidemia e doença aterosclerótica prematura em crianças e adultos. Como as manifestações da LAL-D podem assemelhar-se àquelas observadas em outras doenças mais comuns, o atraso no diagnóstico não é incomum. A doença de Wolman é um fenótipo LAL-D de início precoce que geralmente é fatal no primeiro ano de vida, porém a forma mais comum da doença pode ocorrer em todas as idades. Recentemente, a terapia de reposição enzimática (ERT) para LAL-D tornou-se disponível.
RELATO DE CASO: Paciente, 12 anos, sexo masculino, foi encaminhado para investigação de hepatomegalia. Inicialmente diagnosticado como portador de doença de Niemann-Pick tipo C (NPC), não apresentava mutações confirmatórias dessa doença. Posteriormente, com dosagem enzimática da lipase ácida lisossomal em papel-filtro foi redirecionado o diagnóstico para LAL-D (com confirmação em dosagem enzimática em leucócitos e fibroblastos).
DISCUSSÃO: Apesar de LAL-D ser considerada uma rara e incomum causa de doença hepática, estudos recentes apontam para uma prevalência maior dessa doença. Diagnóstico precoce é fundamental, visto já ser disponível tratamento específico para essa doença. Cerca de metade dos pacientes com LAL-D falecem antes dos 21 anos de idade na ausência de tratamento adequado.
Palavras-chave:
Encefalopatias metabólicas congênitas, Fígado gorduroso, Hepatomegalia, Diagnóstico.
ABSTRACT
INTRODUCTION: Lysosomal acid lipase deficiency (LAL-D) is a lysosomal storage disorder involved in cholesterol ester metabolism. It is a poorly understood genetic cause of cirrhosis, dyslipidemia and premature atherosclerotic disease in children and adults. As the manifestations of LAL-D may resemble those observed in other more common diseases, delayed diagnosis is not uncommon. Wolman disease is an early-onset LAL-D phenotype that is usually fatal in the first year of life, but the most common form of the disease may occur at all ages. Recently, enzyme replacement therapy (ERT) for LAL-D has become available.
CASE REPORT: A 12-year-old male patient was referred for hepatomegaly. Initially diagnosed as having Niemann-Pick type C disease (NPC), it did not present confirmatory mutations of this disease. Later, with enzymatic dosage of lysosomal acid lipase on filter paper, the diagnosis was redirected to LAL-D (with confirmation in enzymatic dosage in leukocytes and fibroblasts).
DISCUSSION: Although LAL-D is considered a rare and uncommon cause of liver disease, recent studies point to a higher prevalence of this disease. Early diagnosis is essential, since specific treatment for this disease is already available. About half of patients with LAL-D die before age 21 in the absence of adequate treatment.
Keywords:
Metabolism. Inborn errors. Fatty liver. Hepatomegaly. Diagnostic tests. Routine.
INTRODUÇÃO
A deficiência de lipase ácida lisossômica (LAL) consiste em uma doença metabólica hereditária, pertencente a um grupo seleto de erros inatos do metabolismo denominado doenças de depósito lisossômico (LSD)1.
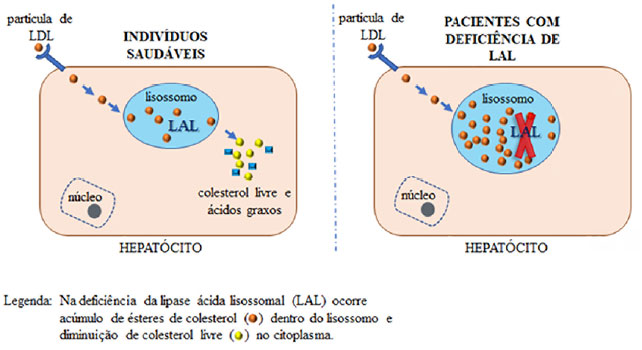
A enzima LAL é responsável por degradar um tipo particular de gordura conhecida como éster de colesterol (colesterol esterificado combinado a triglicerídeos) (Figura 1), e a deficiência dessa enzima acarreta acúmulo progressivo dos ésteres de colesterol em órgãos vitais como fígado, baço, glândulas suprarrenais, sistema hematopoiético, intestinos e na parede de vasos sanguíneos, proporcionando alguns sinais e achados frequentes nessa enfermidade como hepatopatia e dislipidemia2.
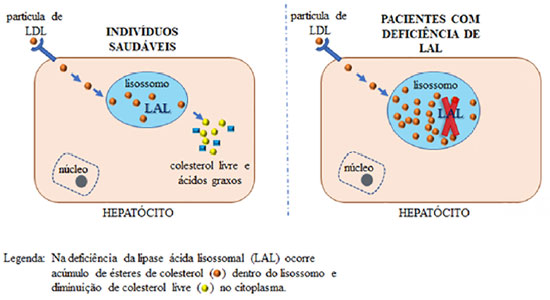 Figura 1.
Figura 1. Representação esquemática comparando célula saudável e célula com deficiência de LAL. Fonte: Os autores (2021).
A forma de manifestação precoce da deficiência de LAL, quando manifestada nos primeiros meses de vida, foi nomeada como doença de Wolman3. Nesses casos, trata-se de uma forma mais agressiva dessa enfermidade, em que os pacientes apresentam diarreia intratável, dificuldade de ganho pôndero-estatural, calcificação de adrenais, sendo rapidamente fatal até o 6º mês de vida3.
Já a forma considerada "tardia" da deficiência de LAL era denominada de CESD (Cholesteryl Ester Storage Disease - doença de depósito de ésteres de colesterol), apresentando os primeiros sintomas usualmente após os primeiros 6 meses de vida até a idade adulta3. Nessa forma mais comum da doença, fibrose hepática, cirrose, insuficiência hepática, acidente vascular cerebral em idade precoce, risco aumentado de doenças cardiovasculares (devido à aterosclerose prematura) são as manifestações clínicas mais frequentes1.
Devido à grande variabilidade clínica na apresentação da deficiência de LAL, o atraso diagnóstico é comum e muitos pacientes podem ser diagnosticados de forma incorreta como portadores de "fibrose/cirrose hepática idiopática" ou "esteatose hepática não alcoólica criptogênica"4.
Em relação ao tratamento, até recentemente ainda não existem terapias específicas para a deficiência de LAL, alguns fármacos para controle da dislipidemia (como as estatinas) não trouxeram resultados eficazes1, porém, através de estudos em modelos animais com administração de uma forma recombinante da enzima LAL (sebelipase), foi possível realizar-se ensaios clínicos em seres humanos que culminaram com estudo clínico de fase III em 2015 cujos resultados demonstraram diminuição significativa nas transaminases, gordura hepática, LDL, triglicerídeos, e aumento do HDL1. Posteriormente, órgãos como o FDA (Food and Drugs Administration) e o EMA (European Medicines Agency) aprovaram a terapia com reposição enzimática (TRE) para a deficiência de LAL (administrada por via intravenosa a cada 15 dias)1.
Por se tratar de uma terapia relativamente recente, ainda não existe acompanhamento desse tratamento em longo prazo, o que será importante para determinar se a progressão da enfermidade para insuficiência hepática pode ser garantida nos pacientes que iniciaram a terapia antes que danos hepáticos permanentes tenham se instalado1. Torna-se, portanto, importante que os pacientes em terapia com a enzima recombinante fiquem em acompanhamento clínico contínuo para se observar os efeitos e limitações dessa terapia em um período de tempo mais prolongado (e no contexto clínico do dia a dia, fora de um ensaio clínico controlado)1.
Relatamos aqui caso de primeiro paciente brasileiro diagnosticado com deficiência de lipase ácida lisossomal por meio da técnica de dosagem enzimática em papel-filtro.
RELATO DE CASO
Paciente, sexo masculino, 12 anos, primeiro e único filho de pais jovens e não consanguíneos, foi encaminhado para investigação de colestase prolongada com hepatomegalia.
Seus antecedentes gestacionais não revelaram intercorrências. Nasceu de parto cesáreo, a termo, p=2940g, comprimento de 46,5cm, perímetro cefálico de 33cm, perímetro torácico de 32cm, com APGAR 8/9. Paciente evoluiu com icterícia neonatal prolongada (inicialmente por suspeita de incompatibilidade ABO), necessitando realizar exanguíneo transfusão. Obteve alta no quinto dia de vida.
Não apresentou alterações de marco de neurodesenvolvimento, andando com cerca de 1 ano de idade, elaborando frases com 1 ano e 6 meses. De antecedentes mórbidos, paciente apresenta dislipidemia com diminuição do HDL, aumento do LDL, dos triglicerídeos e do colesterol total, além de aumento persistente das transaminases.
Aos 3 anos, foi observado aumento de baço e fígado, sendo encaminhado para realização de biópsia hepática, cujo resultado foi compatível com uma doença metabólica hereditária. Fez-se investigação para outras doenças metabólicas que cursam com alteração hepática, porém resultados foram considerados normais, com exceção de teste de filipin para doença de Niemann-Pick tipo C cujo padrão de fluorescência perinuclear foi considerado compatível com diagnóstico dessa enfermidade. Como esse teste pode ter alteração em outras enfermidades que interfiram no metabolismo do colesterol intracelular, a confirmação diagnóstica faz-se necessariamente com análise genético-molecular. Sequenciamento dos genes NPC1 e NPC2, contudo, foi normal, afastando essa hipótese.
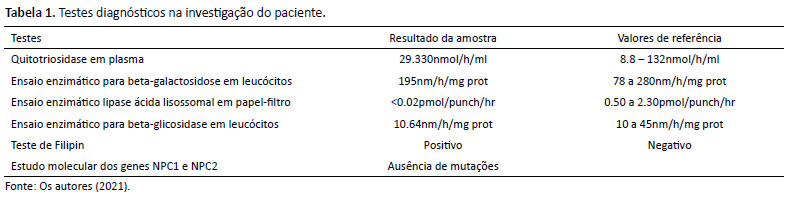
Diante da não elucidação diagnóstica, paciente ficou em avaliação de rotina até realizar teste enzimático para deficiência de lipase ácida lisossomal em Glasgow na Escócia, pois, à época, não havia esse ensaio enzimático disponível ainda no Brasil (Tabela 1). Estudo enzimático em papel-filtro confirmou diagnóstico de deficiência de lipase ácida lisossomal (LAL) no paciente, posteriormente repetido em leucócitos e fibroblastos corroborando resultados obtidos previamente em papel-filtro.
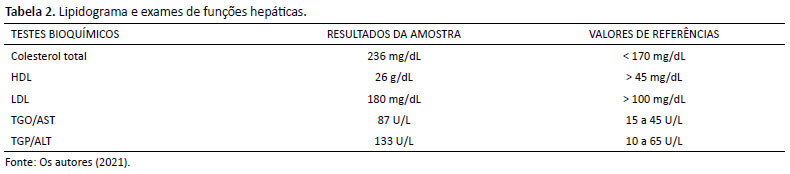
Posteriormente ao diagnóstico enzimático, iniciou-se acompanhamento mais rigoroso de dislipidemia. Em seus exames, há alteração no colesterol LDL e HDL, alto e baixo, respectivamente, e nas enzimas hepáticas TGO e TGP, ambas aumentadas (Tabela 2).
Após o diagnóstico, introduziu-se uma dieta hipolipídica e rica em fibras, visando ao controle de dislipidemia, evitando-se gorduras poli-insaturadas, alimentos industrializados e excesso de carboidratos.
Seu exame genético-clínico mais recente revelou: estatura e peso entre os percentis 25 e 50; perímetro cefálico entre os percentis 50 e 97, sem fácies infiltrada, presença de hepatoesplenomegalia, reflexos osteotendíneos grau 2 em membros inferiores e superiores, ausência de sinais de compressão medular e de apraxia oculomotora, paciente eumétrico e com eudiacocinesia; sensibilidade profunda preservada.
DISCUSSÃO
O diagnóstico de deficiência de lipase ácida lisossomal (LAL-D) é desafiador, pois compartilha das mesmas características clínicas e laboratoriais que outras condições, como a esteato-hepatite não alcoólica, hiperlipidemia familiar combinada e outros problemas metabólicos mais comuns3. A LAL-D apresenta-se em um espectro clínico desde a infância até a idade adulta (Tabela 3), semelhante a outras doenças de armazenamento lisossômico3.
Aqueles que manifestaram sintomas no primeiro ano de vida eram historicamente conhecidos como doença de Wolman, a qual possui uma apresentação rapidamente progressiva, sendo descrita pela primeira vez em 1956, visto que, enquanto os que desenvolviam as manifestações clínicas na infância eram descritos como doença de armazenamento dos ésteres de colesterol (CESD) ou mais modernamente apenas como LAL-D5. Na doença de Wolman, os lactentes afetados foram diagnosticados com desnutrição grave, calcificações adrenais, hepatoesplenomegalia e morte nos primeiros meses de vida. Em contraste, a CESD é vista como tendo um espectro clínico mais amplo com apresentações que podem surgir desde a infância até a idade adulta apresentando-se, por vezes, como cirrose criptogênica5.
Pacientes com LAL-D, muitas vezes, podem ser diagnosticados tardiamente ou mesmo incorretamente. Uma vez que um tratamento específico está agora disponível para tal enfermidade, o diagnóstico de LAL-D deve figurar no diagnóstico diferencial de lactentes com síndrome de má absorção grave ou colestase prolongada neonatal de causa desconhecida5.
Além disso, a fibrose hepática "idiopática" e cirrose criptogênica em crianças ou adultos devem desencadear um maior nível de suspeita para o rastreio de LAL-D. Sendo assim, as imagens diagnósticas, como a ultrassonografia e a biópsia hepática, são relevantes para o diagnóstico (embora possam ser inespecíficas em muitos pacientes) e demonstram alterações na morfologia hepática, que podem ser sugestivas de LAL-D, como esteatose microvesicular com envolvimento de células de Kupffer, fibrose e deposição de cristais de éster de colesterol (quando realizada microscopia eletrônica para análise do tecido hepático biopsiado).
Como visto nos achados do presente caso, a doença se manifesta como uma doença idiopática microvesicular hepatosteatose. Assim, após essa análise, é possível realizar diagnóstico presuntivo de LAL-D6.
O teste específico para o diagnóstico de deficiência de LAL é baseado na atividade enzimática em leucócitos do sangue periférico, cultura de fibroblastos da pele e, mais recentemente, na análise enzimática feita em gota de sangue seco coletada em papel-filtro, permitindo o reconhecimento rápido e o material de transporte fácil e prontamente disponível para testes. O teste enzimático em papel-filtro pode ser utilizado para a triagem seletiva de pacientes apresentando, por exemplo, esteatose hepática não alcoólica (especialmente em pacientes não obesos) e hepatomegalia/esplenomegalia sem etiologia definida (Tabela 4).
Por causa da natureza hereditária da doença, os pacientes diagnosticados e suas famílias têm direito a aconselhamento genético. Nesses casos, o diagnóstico pré-natal e a determinação do conhecimento do portador das mutações no LIPA aceleram e simplificam bastante o processo de diagnóstico.
O paciente em questão foiinicialmente diagnosticado como portador de doença de Niemann-Pick tipo C (NPC) e, posteriormente, com dosagem enzimática da lipase ácida lisossomal em papel-filtro foi realizado o correto diagnóstico de LAL-D (com confirmação também em análise enzimática em leucócitos e fibroblastos, após realização do teste em papel-filtro). Diante disso, verifica-se que a utilização da análise enzimática em papel-filtro como forma de triagem tem diversas vantagens, como o fácil transporte das amostras, podendo ser enviado via correio em envelope convencional, sem necessidade de refrigeração7, fácil armazenagem das amostras, com atividade preservada até seis meses a temperatura ambiente e durante dois anos a 4 °C8-10; menor volume de reação, economizando reagentes e quantidade de amostra, tornando-se mais barato que o uso de leucócitos, plasma ou fibroblastos11,12.
O tratamento consiste, para crianças e adultos com deficiência de LAL (CESD), em medicamentos redutores de colesterol ("estatinas" e/ou ezetimibe) para controle do colesterol "ruim" (LDL) no sangue. Embora esses medicamentos possam reduzir o colesterol, não demonstraram interferir na doença de base nem prevenir as graves complicações hepáticas. Além disso, há evidências de que, em alguns casos, essas drogas possam mascarar a progressão da doença4.
A terapia de reposição enzimática já foi aprovada na União Europeia e nos Estados Unidos desde 2015 e tem sido utilizada com grande eficácia. Dessa forma, o diagnóstico precoce dessa condição mórbida é crítico e de suma importância para o melhor manejo terapêutico do paciente. Embora os achados histopatológicos hepáticos possam sugerir essa doença ou levar a outros diagnósticos diferenciais, atualmente a mensuração da atividade enzimática da LAL tornou-se ferramenta fundamental no diagnóstico precoce dessa enfermidade, evitando diagnósticos errôneos (como no caso do paciente do relato) e permitindo iniciar-se terapêutica específica antes que lesões irreversíveis em órgãos-alvo ocorram.
REFERÊNCIAS
1. Balwani M, Breen C, Enns GM, Deegan PB, Honzík T, Jones S et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology 2013;58(3):950-7.
2. Wierzbicka-Rucińska A, Jańczyk W, Ługowska A, Lebensztejn D, Socha P. Diagnostic and therapeutic management of children with lysosomal acid lipase deficiency (LAL-D). Review of the literature and own experience. Dev Period Med 2016;20(3):212-5.
3. Tommaso AMAD, Barra FF de C, Hessel G, Moreno CA, Giugliani R, Escanhoela CAF. Importance of liver biopsy in the diagnosis of lysosomal acid lipase deficiency: a case report. Rev Paul Pediatr 2018;36(1):113-6.
4. Bay L, Canero Velasco C, Ciocca M, Cotti A, Cuarterolo M, Fainboim A, et al. Liver disease and dyslipidemia as a manifestation of lysosomal acid lipase deficiency (LAL-D). Clinical and diagnostic aspects, and a new treatment. An update. Arch Argent Pediatr. 2017 Jun;115(3):287-93.
5. Kuloglu Z, Kansu A, Selbuz S, Kalaycı AG, Şahin G, Kirsaclioglu CT, et al. The frequency of lysosomal acid lipase deficiency in children with unexplained liver disease. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):371-6.
6. Botero V, Garcia VH, Gomez-Duarte C, Aristizabal AM, Arrunategui AM, Echeverri GJ, et al. Lysosomal acid lipase deficiency, a rare pathology: the first pediatric patient reported in Colombia. Am J Case Rep. 2018 Jun;19:669-72.
7. Rodrigues MDB, de Oliveira AC, Müller KB, Martins AM, D'Almeida V. Chitotriosidase determination in plasma and in dried blood spots: a comparison using two different substrates in a microplate assay. Clin Chim Acta. 2009;406(1-2):86-8.
8. Gasparotto N, Tomanin R, Frigo AC, Niizawa G, Pasquini E, Blanco M, et al. Rapid diagnostic testing procedures for lysosomal storage disorders: alpha-glucosidase and beta-galactosidase assays on dried blood spots. Clin Chim Acta. 2009 Apr;402(1-2):38-41.
9. de Castilhos CD, Mezzalira J, Goldim MP, Coelho JC. Influence of pre-analytical factors on α-galactosidase A, arylsulfatase B and α-glucosidase activities measured on dried blood spots on filter paper. Clin Biochem. 2011 Jul;44(10-11):922-6.
10. Castilhos CD, Mezzalira J, Goldim MPS, Werlang FG, Coelho JC. Effect of sample collection, temperature and time of storage on β-galactosidase and total hexosaminidase activities in dried blood collected on filter paper. Clin Chem Lab Med. 2011;49(8):1299-302.
11. Civallero G, Michelin K, de Mari J, Viapiana M, Burin M, Coelho JC, et al. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin Chim Acta. 2006 Oct;372(1-2):98-102.
12. Benevides GN, Miura IK, Person NC, Pugliese RPS, Danesi VLB, Lima FR, et al. Lysosomal acid lipase deficiency in Brazilian children: a case series. J Pediatr (Rio J). 2019 Sep-Oct;95(5):552-8.
1. Centro Universitário Estácio de Ribeirão Preto, Genética Clínica - Ribeirão Preto - São Paulo - Brasil
2. CPDP - Centro Paulista de Diagnóstico, Pesquisa e Treinamento - Medicina Diagnóstica, Genética Clínica - Ribeirão Preto - São Paulo - Brasil
3. Laboratório DLE, Bioquímica Genética - Rio de Janeiro - Rio de Janeiro - Brasil
4. Hospital de Clínicas de Porto Alegre, UFRGS, Bioquímica Genética - Porto Alegre - Rio Grande do Sul - Brasil
5. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - São Paulo - Brasil
Endereço para correspondência:
Charles Marques Lourenço
Centro Universitário Estacio de Ribeirão Preto
Rua Abrahão Issa Halach, nº 980, Ribeirânia
Ribeirão Preto, SP, Brasil. CEP: 14096-160
E-mail: charlesgenetica@gmail.com
Data de Submissão: 14/05/2021
Data de Aprovação: 13/06/2021