Artigo Original
-
Ano 2018 -
Volume 8 -
Supl.1
Deficiência intelectual na criança
Inetellectual disabilities in children
Regina Célia Beltrão Duarte
RESUMO
O retardo mental, denominado mais recentemente como deficiência intelectual (DI), é um transtorno neurológico comum na infância e adolescência. Os déficits encontrados envolvem a cognição e o comportamento adaptativo, com início antes dos 18 anos. Inúmeras são as etiologias, desde fatores pré-natais, perinatais e pós-natais até os casos de origem genética. Muitas síndromes genéticas estão associadas. Não há tratamento específico. A assistência geral requer a participação de vários profissionais, tendo o pediatra como coordenador dos diversos encaminhamentos para outras especialidades, conforme as necessidades do quadro clínico.
Palavras-chave:
Deficiência Intelectual, Prevalência, Etiologia Criança.
ABSTRACT
Mental retardation, more appropriately known as intellectual disability (ID), is a common neurologic condition in childhood and adolescence. The clinical deficits involve cognition and adaptive behavior, and its onset occurs before 18 years of age. There is a number of etiologies, ranging from prenatal, perinatal and postnatal factors to cases of genetic origin. Many genetic syndromes are associated with ID. There is no specific treatment. General care requires the participation of several professionals, while the pediatrician acts as the coordinator of referrals to a range of specialties, according to the needs of the clinical picture.
Keywords:
Intellctual Disability, Etiology Prevalence, Child.
INTRODUÇÃO
O retardo mental é um dos transtornos neuropsiquiátricos mais comuns em crianças e adolescentes1.
O conceito de deficiência intelectual foi modificado por vários anos com inúmeras definições e terminologia como oligofrenia, retardo mental e deficiência mental. Segundo Krynskis e colaboradores (1969): a deficiência intelectual possui um espectro complexo de quadros clínicos decorrentes de diferentes etiologias e se caracteriza pelo desenvolvimento intelectual insuficiente2.
A definição de retardo mental está baseada nos sistemas de classificação relatados a seguir3,4.
O termo deficiência intelectual (DI) corresponde ao retardo mental no CID-10 (Código Internacional de Doenças), que utiliza a pontuação do QI (quociente de inteligência) como aspecto mais importante para defini-la, de acordo com o seguinte sistema de classificação:
Retardo mental leve (F70),
Retardo mental moderado (F71),
Retardo mental grave (F72),
Retardo mental profundo (F73).
Apesar de inúmeras críticas ao esquema de normatização e avaliação da inteligência, o critério estatístico para fins práticos de classificação do retardo mental proposto pela Organização Mundial de Saúde (OMS) é demonstrado a seguir:
A outra classificação é o Manual Diagnóstico e Estatístico de Transtornos Mentais (DSM), que se encontra na quinta revisão.
No DSM-5, o retardo mental é substituído por deficiência intelectual, com início no período do desenvolvimento, com déficits funcionais tanto intelectuais quanto adaptativos nos domínios conceitual, social e prático5,6.
Os déficits nas funções intelectuais são confirmados pela avaliação clínica e testes de inteligência padronizados e individualizados, e realizados em crianças a partir de 5 anos, enquanto os déficits adaptativos limitam o funcionamento de uma ou mais atividades diárias, comprometendo a comunicação e o aspecto social, com repercussões nos diversos ambientes: casa, escola e trabalho5,6.
A deficiência intelectual (DI) é uma condição clínica caracterizada por limitações evidentes no funcionamento intelectual e no comportamento adaptativo, este último expresso como habilidades adaptativas conceituais, sociais e práticas e as limitações devem estar presentes antes dos 18 anos7.
A DI deve ser diagnosticada após os 5 anos, quando é possível mensurar a inteligência por meio de testes de QI. Antes disso, o termo bastante utilizado, apesar de muito controverso, é o atraso no desenvolvimento neuropsicomotor8.
Uma pontuação do quociente de inteligência (QI) abaixo de 68 na escala de Standford-Binet ou abaixo de 70 no teste de Wechsler define a presença de disfunção intelectual9.
DEFINIÇÃO DE INTELIGÊNCIA8,9
É uma habilidade mental geral. Inclui raciocínio, planejamento, resolução de problemas, pensamento abstrato, compreensão de ideias complexas, aprendizagem rápida e a partir de experiências2,9.
O conceito de quociente intelectual foi estabelecido pela primeira vez por Binet e Simon a partir dos dados que relacionavam a idade cronológica e a idade mental.
No DSM-5, o retardo mental é substituído por deficiência intelectual. O critério de QI não é a característica central do diagnóstico. O diagnóstico é baseado no nível das funções adaptativas nos domínios social, conceitual e habilidades práticas. Há 4 níveis de severidade: leve, moderada, grave e profunda, baseados nos três domínios do comportamento adaptativo6.
COMPORTAMENTO ADAPTATIVO10
O comportamento adaptativo é a reunião de habilidades conceituais, sociais e práticas que foram aprendidas pelas pessoas para funcionarem em suas vidas diárias (Quadro 1)10.
EPIDEMIOLOGIA11,12
A taxa de prevalência na população em geral é de 1% a 3% de acordo com estudos epidemiológicos. É mais frequente no sexo masculino e nas classes socioeconômicas menos favorecidas.
FATORES ETIOLÓGICOS DO RETARDO MENTAL8,13,14
Inúmeros fatores, genéticos e teratogênicos, causam a DI, entre estes estão o uso do álcool na gravidez, agentes infecciosos e defeitos congênitos do sistema nervoso central (SNC), que apesar de serem congênitos, não significa que sejam geneticamente determinados (Quadro 2). Ao considerarmos as causas genéticas responsáveis pela deficiência intelectual, temos as aberrações cromossômicas numéricas ou estruturais, microdeleções ou microduplicações, defeitos gênicos (monogênicos ou oligogênicos) ou casos de deficiência intelectual resultante da combinação de fatores genéticos e ambientais, como acontece nas doenças de herança multifatorial (Quadro 3). Os erros inatos do metabolismo representam 1% a 5% dos casos de DI.
As causas de DI geneticamente determinadas podem ocorrer de forma isolada (não sindrômica) ou associada a outros sinais e sintomas físicos (sindrômicas), sugerindo um quadro específico; ou seja, além da DI, o paciente apresenta um quadro que caracteriza uma síndrome, por exemplo a síndrome de Down ou trissomia do 21. De qualquer maneira, sendo a DI sindrômica ou não, há um impacto negativo no paciente acometido e nos seus familiares.
Vários estudos demonstraram os principais fatores etiológicos da DI. Em um estudo realizado na Colômbia, 239 pacientes com DI foram avaliados e as principais causas encontradas foram os fatores pré-natais (infecções e prematuridade) e perinatais (hipóxia e hiperbilirrubinemia, HIV e hipotireoidismo congênito) e pós-natais (infecções do SNC, traumatismo cranioencefálico). Neste estudo, o diagnóstico definitivo mais frequente foram causas ambientais (36,4%); a segunda causa, genética (23,8%); a terceira, multifatorial (4,2%); e sem diagnóstico definitivo (23,8%)13.
Essas proporções variam muito conforme a população estudada, a metodologia do estudo, a época do estudo e a disponibilidade de exames genéticos13.
A DI é causada por inúmeros fatores ambientais e genéticos, porém em 55% a 60% dos casos as causas são indefinidas16.
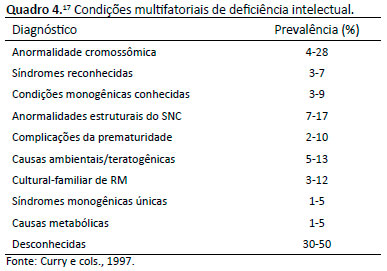
Em outros estudos, as causas genéticas de DI foram identificadas entre 17% a 40% dos casos examinados, enquanto as causas ambientais, malformações do SNC e condições multifatoriais foram responsáveis por quase 30% dos casos (Quadro 4)17.
A seguir, algumas síndromes que cursam com DI:
SÍNDROME ALCOÓLICA-FETAL (SAF)OU TRANSTORNOS DO ESPECTRO ALCOÓLICO FETAL18
É observada na prole de gestantes que consumiram bebidas alcoólicas. O quadro clínico se caracteriza por DI, microcefalia, retardo do crescimento pré e pós-natais e dismorfismos faciais (pregas epicânticas, nariz curto, pequena abertura dos olhos, fácies planas), anomalias renais, cardiopatia e baixa estatura.
Apesar de SAF estar presente em filhos de mães etilistas, não há uma dose segura de álcool para ser ingerida na gravidez; dessa forma é aconselhável à gestante não beber durante toda a gravidez. O álcool interfere no processo de maturação neuronal, nas etapas de migração e mielinização e favorece a produção de radicais livres.
ERROS INATOS DO METABOLISMO (EIM)19
São doenças genéticas crônicas com manifestações clínicas em qualquer idade. As manifestações clínicas dos EIM são inespecíficas e se confundem com doenças mais comuns, fator que contribui para o diagnóstico tardio. Os pacientes afetados podem apresentar episódios recorrentes de descompensação metabólica com alterações em múltiplos órgãos, sintomas neurológicos agudos ou progressivos e atraso no desenvolvimento neuropsicomotor, além de apresentarem frequentemente problemas comportamentais e no aprendizado. Alguns desses EIM podem ser detectados no “teste do pezinho”, como a fenilcetonúria. Outros testes podem ser realizados em laboratório particular para detecção de algumas doenças, como doença do xarope de bordo, galactosemia e frutosemia, entre outras.
Inúmeros estudos relacionam a DI com os erros inatos do metabolismo. Com base em bases de dados internacionais, há mais de 7.300 estudos relacionando os temas “erros inatos do metabolismo” e DI. Uma das doenças que requer um diagnóstico precoce com tratamento imediato é a fenilcetonúria. Do ponto de vista neurológico, os sinais e sintomas do paciente com fenilcetonúria apresentam as manifestações clínicas já nos primeiros meses de vida e incluem irritabilidade e atraso no desenvolvimento neuropsicomotor, além de sintomas posteriores como dificuldade no aprendizado e sintomas comportamentais, como no transtorno do déficit de atenção e hiperatividade (TDAH). A principal manifestação neurológica da fenilcetonúria é a deficiência intelectual. A fenilcetonúria foi o primeiro EIM em que foi identificada a relação entre um aumento da substancia tóxica e o desenvolvimento de DI. O diagnóstico precoce e a implementação imediata da dieta restrita em fenilalanina continua sendo o tratamento mais eficaz para os fenilcetonúricos, sendo capaz de prevenir os danos neurológicos entre eles a DI.
SÍNDROME DO X FRÁGIL (SXF)15,16,20
É a segunda causa de deficiência intelectual de origem genética. Acomete 1 em cada 4.000 a 6.000 nascimentos do sexo masculino e 1 a cada 8.000 a 9.000 do sexo feminino.
O achado molecular é uma mutação que leva à supressão da transcrição do gene FMR1, localizado na região Xq27.3, caracterizada pela expansão de trinucleotídeos CGG.
Na população normal, o número de repetições é de 5 a 44 trincas CGG nessa região. Os pacientes com 50 até 200 repetições são considerados portadores da pré-mutação e aqueles com mais de 200 repetições são considerados portadores da mutação completa para essa síndrome.
O quadro clínico de um paciente do sexo masculino com mutação completa apresenta DI geralmente moderada, timidez com fixação pobre do olhar, hiperatividade, aversão ao toque. Em virtude do quadro, muitos desses pacientes estão dentro do espectro autista. Apresentam ainda orelhas grandes, face alongada, macro-orquidismo. hiperextensibilidade articular e crises epilépticas.
Pacientes do sexo masculino com a pré-mutação na idade adulta podem apresentar síndrome de tremor e ataxia associada a X frágil. No sexo feminino com a mutação completa, apresentam DI leve e timidez, podendo evoluir para a menopausa precoce. Aproximadamente 21% dos pacientes do sexo feminino com pré-mutação evoluem para a menopausa antes dos 40 anos, situação clínica conhecida como falência ovariana precoce. O diagnóstico dessa síndrome é a investigação da mutação do gene FMR1 e o método é por meio do PCR ou técnica de Southern Blotting (padrão-ouro).
O American College of Medical Genetics recomenda a análise do gene FMR1 para: 1) pessoas de ambos os sexos que apresentem DI ou transtorno do espectro autista, especialmente quando ocorrem características físicas e comportamentais da SXF, história familiar da síndrome do X frágil ou parente com DI de causa desconhecida; 2) pessoas que buscam conhecer o risco para a prole, porque têm história familiar de síndrome do X frágil ou de DI de causa desconhecida; 3) mulheres com insuficiência ovariana, especialmente se há história familiar de menopausa precoce, casos de DI de origem desconhecida e casos de SXF; 4) pessoas com ataxia/tremor de manifestação tardia, cuja causa é desconhecida, ou casos de DI de causa desconhecida ou casos de história familiar de SXF.
A SXF tem herança ligada ao cromossomo X e afeta aproximadamente 2,5% dos meninos e 1% das meninas com DI. Como essa síndrome é hereditária, o diagnóstico dessas crianças é de fundamental importância para a orientação dos pais quanto ao risco de repetições da SXF em outra criança que venha apresentar. A hipótese da SXF deve ser considerada em crianças com DI ou autismo, principalmente associada com alterações físicas e comportamentais, ou a ocorrência na família materna de SXF ou DI de causa desconhecida.
Inúmeras doenças genéticas são responsáveis pela DI, e a descrição de cada doença seria exaustiva, sendo que vale lembrar que vários fatores ambientais e genéticos são responsáveis pela DI; assim como uma grande maioria, em quase 50% a 60% dos casos de DI, permanecem até o momento sem causa definida.
SÍNDROME DE DOWN15
A síndrome de Down ou trissomia do 21 é a causa mais comum de DI1. É considerada a anomalia cromossômica mais frequente nos seres humanos comparada com outras trissomias, como dos cromossomos 18 e 13. A incidência é estimada em 1:800 nascidos vivos, acometendo todas as raças e classes sociais. As crianças afetadas apresentam um QI médio de 50 e o diagnóstico é, geralmente, suspeitado a partir das manifestações clínicas como hipotonia, braquicefalia, macroglossia, fácies plana, fissuras palpebrais oblíquas, língua protusa, prega palmar única, háluces mais distantes dos demais artelhos e malformações de múltiplos órgãos e/ou de sistemas.
QUADRO CLÍNICO DA DEFICIÊNCIA INTELECTUAL21-26
O diagnóstico precoce da DI contribui para uma intervenção mais antecipada com identificação das habilidades, melhor aceitação da criança na comunidade e melhora da ansiedade dos pais. A maioria das crianças, quando lactentes, apresentam atraso no desenvolvimento neuropsicomotor ou dismorfismos. Não há alterações físicas específicas nos casos de deficiência intelectual, porém os dismorfismos encontrados na criança podem representar o primeiro sinal de alguma DI. Em lactentes, é observada uma falta de resposta a estímulos visuais e auditivos, alterações posturais como hipotonia ou hipertonia e dificuldades na alimentação. A DI grave é geralmente identificada por volta dos 3 anos. No caso de DI leve, o diagnóstico é mais tardio e permanece nos primeiros anos escolares, nos quais a criança não consegue acompanhar as demandas acadêmicas ou sociais típicas da idade; e, posteriormente, são observadas as limitações da criança conforme as demandas escolares oferecidas. Em adolescentes, a DI leve não é facilmente identificada e muitos casos são diagnosticados como transtornos de aprendizagem (p. ex., dislexia) ou mascaram o comportamento, sendo rotulados de “agressivos” ou “incompetentes”.
No caso de síndrome genética, como a de Down, os sinais clínicos clássicos demonstram a etiologia da DI, mas outros sinais indiretos também levam a suspeita clínica, como no caso da micro ou macrocefalia. Outras patologias geralmente acompanham a DI, como a paralisia cerebral, epilepsia, bebês hipotônicos, autismo, entre outras. Outra causa de deficiência intelectual, no caso de meninas, é a síndrome de Rett, em que os primeiros sintomas da síndrome ocorrem após 6 a 18 meses do desenvolvimento normal, quando a criança apresenta perda da fala, movimentos estereotipados das mãos, crises epilépticas e alterações respiratórias, evoluindo para alterações motoras como no caso do comprometimento da marcha. O gene implicado na síndrome de Rett é o MECP2.
As crianças com DI grave ou profunda normalmente necessitam de atenção mais precocemente, pois apresentam comprometimento clínico, algumas com alterações dismórficas, distúrbios psiquiátricos e de comportamento, e isso leva o clínico a pensar em um atraso cognitivo global.
Os pacientes com DI leve não são rapidamente diagnosticados, sendo encaminhados para avaliações com vários profissionais, quando, então, passam a apresentar problemas acadêmicos. Não é tarefa fácil diferenciar DI leve com transtornos de aprendizagem e, geralmente, as queixas mais frequentes dos pais para o pediatra são atraso na fala, baixo rendimento escolar e alterações no comportamento.
Nos transtornos de aprendizagem ocorre um comprometimento significativo na habilidade escolar específica, seja na leitura, escrita ou matemática. Nessa categoria são exemplos a dislexia e a discalculia.
As crianças com DI geralmente têm associações com outros quadros clínicos, como distúrbios visuais, auditivos, ortopédicos, comportamentais e emocionais. Alguns desses distúrbios são detectados mais tardiamente em crianças com DI, e se não forem tratados, esses déficits podem potencialmente afetar o desempenho do indíviduo, sendo algumas vezes mais graves que a própria deficiência intelectual. Os problemas mais frequentes que estão associados à DI são a deficiência motora, crises epilépticas, distúrbios de comportamento e quadros emocionais. Quanto maior a gravidade da deficiência intelectual, maior o número e a gravidade de patologias associadas.
DIAGNÓSTICO DA DEFICIÊNCIA INTELECTUAL: EXAME FÍSICO GERAL E NEUROLÓGICO E PESQUISA DE DISMORFISMOS
Na anamnese: investigar história familiar de doenças neurológicas e de deficiência intelectual, história de consanguinidade entre os pais e na família, nível educacional dos pais, história da gestação e parto, e heredograma. No exame físico: medida do perímetro cefálico, inspeção da pele, exame físico geral e neurológico, e pesquisa das anomalias congênitas (algumas são sutis)14,23.
Os exames mais frequentes utilizados para investigação de DI são os exames de neuroimagem, como tomografia ou ressonância magnética de crânio. Esses exames são solicitados principalmente em casos de microcefalia, macrocefalia, crises epilépticas, atraso no desenvolvimento neuropsicomotor e sinais neurológicos focais27. O exame de ressonância magnética do crânio tem maior sensibilidade quando comparado à tomografia de crânio, exceto na suspeita de infecções congênitas. As principais alterações encontradas na RM de crânio foram a displasia do corpo caloso, persistência do cavum do septo pelúcido e/ou vergae, ventriculomegalia, hipoplasia vermiana, displasias corticais e alargamento do espaço subaracnóideo28,29. Outros exames podem ser solicitados, como eletroencefalograma (EEG) e vídeo-EEG de acordo com o quadro clínico apresentado, pois são exames mais específicos e não são solicitados como exames de triagem. As solicitações dos exames devem ser de acordo com a suspeita clínica, baseadas na história clínica, exame físico e avaliações de outros profissionais especializados.
Os exames solicitados para investigação de DI geralmente englobam avaliação da função da tireoide, investigação de infecções congênitas, (TORCHS e Zika vírus), nível sérico de amônia (casos suspeitos de distúrbios do ciclo da ureia) e da homocisteína (distúrbios da homocisteína). Além disso, triagem para deficiências auditivas e visuais e avaliação do desenvolvimento neuropsicomotor no caso de suspeita do transtorno do espectro autista23.
A frequência de anormalidades cromossômicas em pacientes com DI varia de 4% a 34%; sendo assim, na avaliação de um paciente com DI deve ser solicitado o cariótipo convencional ou exames com técnicas mais complexas que serão descritos posteriormente14,23.
Outros exames menos frequentes, mas que são solicitados no caso de DI: dosagem de ácidos orgânicos e aminoácidos na urina, lactato e piruvato no sangue e líquor, dosagem de creatinoquinase. Poucas doenças metabólicas cursam com DI, sendo a prevalência até 5% segundo alguns estudos. Porém, entre as causas de DI estão os erros inatos do metabolismo e, no caso de suspeita clínica, devem ser solicitados os exames de acordo com a necessidade para elucidação do diagnóstico.
As anomalias cromossômicas e a mutações gênicas podem causar DI. As cromossômicas detectadas nas técnicas convencionais, em geral, resultam em síndromes relacionadas com DI, por causarem alterações em vários genes; entre elas está a trissomia do cromossomo 21 (síndrome de Down), que é a principal causa genética de DI. Recentemente, novas técnicas são utilizadas quando o cariótipo tradicional não revela alterações, entre elas a análise de microarray, utilizada para investigar causas de DI. As microdeleções e microduplicações podem ser responsáveis por 15% das causas de DI. A síndrome de deleção 1p36 é um exemplo de síndrome de microdeleção subtelomérica13,14,17,27.
De um modo geral, a investigação da DI se baseia na história clínica, exames laboratoriais, testes genéticos como o cariótipo com banda G, técnicas mais avançadas para microdeleções ou microduplicações como hibridação genômica comparativa (CGH-array), técnicas como FISH e sequenciamento do exoma14.
Entre os fatores etiológicos da deficiência intelectual estão as causas genéticas, que representam entre 17% a 40% dos casos examinados. O exame de citogenética convencional (cariótipo com banda G) detecta as anomalias cromossômicas. Trata-se de uma técnica de rotina no diagnóstico da DI. É uma técnica sensível e econômica, porém, não detecta segmentos estruturais anômalos muito pequenos. Outro exame capaz de identificar entre 15% a 20% dos casos de DI é a hibridação genômica comparativa em microarranjos de DNA (CGH-array).
O CGH-array é uma técnica de citogenética molecular capaz de identificar alterações cromossômicas. É um exame que permite estudar todo o genoma humano de uma só vez, identificando duplicações, deleções de segmentos cromossômicos, deleções e duplicações. O CGH-array detecta alterações que não são vistas no cariótipo convencional e é o teste oficialmente indicado pela Academia Americana de Genética no estudo de crianças e adultos com suspeita de síndromes genéticas, atraso no desenvolvimento neuropsicomotor, deficiência intelectual e transtorno do espectro autista30.
Todas as alterações detectadas no exame CGH-array são pesquisadas em bancos de dados internacionais de variantes que catalogam os resultados clínicos com a localização de genes e sua função; sendo assim, a interpretação dos resultados requer profissionais treinados, uma vez que muitas das alterações detectadas representam variações de números de cópias sem significado clínico ou de significado clínico desconhecido. O CGH-array é um método que possibilita uma análise refinada de todo o genoma e por esse motivo tem facilitado o diagnóstico e a identificação das bases moleculares de várias alterações genéticas, entre elas a DI30,31.
O advento da técnica de hibridização in situ fluorescente (FISH) permitiu a detecção de regiões específicas. É uma técnica utilizada para detectar anomalias cromossômicas submicroscópicas, como nas síndromes de microdeleções, entre elas a síndrome de Prader-Willi, de Angelman, velocardiofacial, de DiGeorge e de Williams.
A utilização de sondas de DNA para mutações específicas, utilizando a técnica de amplificação de múltiplas sondas dependentes de ligação (MLPA), baseada em uma reação em cadeia de polimerase (PCR), é utilizada para diagnóstico da síndrome de Rett (MECP2)13,14,17,31.
A deficiência intelectual apresenta causas genéticas, porém, mais da metade dos casos ainda são considerados idiopáticos e a aplicação de técnicas de citogenética clássica e molecular tem permitido o diagnóstico preciso em alguns casos. Algumas técnicas são utilizadas para investigação de rearranjos microscópicos em pacientes com DI idiopático, incluindo o cariótipo com bandeamento G, FISH, MLPA e CGH-array33.
Avanços na tecnologia do sequenciamento de nova geração (NGS) permitiram uma redução de tempo e de custos para o sequenciamento do genoma humano. Essa nova tecnologia propiciou o sequenciamento completo do exoma (SCE) e tem sido aplicada na prática clínica, principalmente nos casos de herança monogênica presumida (um único gene envolvido na doença) ou com grande heterogeneidade genética (vários genes responsáveis pelo mesmo quadro clínico)32.
Algumas publicações sugerem que o sequenciamento completo do exoma deve substituir os ensaios por microarranjos como teste de primeira linha. Vale ressaltar que, apesar da associação de diversas abordagens diagnósticas, incluindo ensaios por microarranjos e sequenciamento do exoma, a causa da deficiência intelectual inespecífica continua sem esclarecimento até 60% dos casos34. As mutações raras de ponto ou de novo não são herdadas dos pais e representam uma causa importante de DI, e o sequenciamento do exoma é usado com uma estratégia diagnóstica para a sua detecção34.
Quando se considera um quadro de DI grave no qual há maior chance do achado de um defeito genético causal, a taxa diagnóstica é de 42% após o sequenciamento completo do exoma, que é a técnica mais sensível disponível no momento, visto que sequencia virtualmente todo o genoma humano33.
A técnica do sequenciamento do exoma consiste na coleta de uma amostra de sangue para extração do DNA. Essa técnica permite a análise de cerca de 20 mil genes e vem sendo utilizada nos últimos anos em pesquisa para descoberta de novos genes associados a doenças, constituindo uma importante ferramenta de diagnóstico33. Essa técnica é mais uma ferramenta para definição etiológica em pacientes nos quais algumas técnicas, como cariótipo convencional e CGH-microarray, não identificaram alterações33. O sequenciamento do exoma é um excelente método de diagnóstico para doenças com grande heterogeneidade genética, como a DI32,33,36.
Recentemente, no Brasil, a ANS (Agência Nacional de Saúde Suplementar) incorporou como método complementar de diagnóstico, com diretriz de utilização, o microarray para casos de atraso de desenvolvimento neuropsicomotor ou deficiência intelectual, mas esse teste genético não foi incorporado como exame diagnóstico de primeira linha, sendo exigido o cariótipo convencional, além de outros critérios clínicos35.
No âmbito do SUS (Sistema Único de Saúde), a Política Nacional de Atenção Integral às Pessoas com Doenças Raras, oficializada pela Portaria Nº 199, de 30 de janeiro de 2014, publicada pelo Ministério da Saúde, prevê a realização de microarray para investigação etiológica de condições clínicas que envolvem deficiência intelectual. Tanto o sistema público brasileiro quanto o privado ainda não consideram a possibilidade de utilizar o sequenciamento completo do exoma como exame diagnóstico de forma sistemática. De um modo geral, esse teste tem sido custeado pela família ou, em alguns casos, via judicialização35.
O exoma refere-se a um conjunto de éxons presentes no genoma de grande parte dos seres vivos. O exoma é uma diminuta porção do genoma humano que corresponde ao conjunto de todos os éxons do genoma, isto é, a porção codificante do genoma. O exoma corresponde a menos de 2% do genoma humano, mas é nessa pequena porção do DNA que se concentra a maioria das mutações com potencial patogênico e que são responsáveis pelas doenças geneticamente determinadas35.
O sequenciamento completo do exoma (SCE) tem suas limitações, pois é um exame projetado para avaliar somente éxons e, portanto, as alterações que estiverem fora dos éxons e possam ser responsáveis pelo quadro clínico da DI não serão analisadas por essa técnica. O SCE é uma análise de sequência das bases de DNA, o que significa que alterações no número de cópias (microdeleções, microduplicações), alterações estruturais dos cromossomos, alterações epigenéticas (metilação, acetilação), não podem ser confirmadas nessa análise, necessitando de confirmações por outras técnicas disponibilizadas como cariótipo em banda G, CGH-microarray, FISH e MLPA.
TESTES NEUROPSICOLÓGICOS37
O diagnóstico de deficiência intelectual necessita ser complementado pelos testes neuropsicológicos com a aplicação de testes individualizados de inteligência e comportamento adaptativo. O teste mais utilizado em lactentes é a Escala de Desenvolvimento do Lactente de Bayley, que avalia a linguagem, habilidades na resolução de problemas visuais, comportamento e habilidades motoras. Os testes utilizados em crianças acima de 3 anos são as escalas de Wechsler, e a WPPSI-III é usada para crianças com idade de 3 a 7 anos. A Escala de Wechsler de Inteligência para Crianças - 4ª edição (WISC-IV) é usada em crianças com idade mental superior a 6 anos.
O teste mais comumente utilizado para o comportamento adaptativo é a Escala de Comportamento Adaptativo de Vineland, que envolve entrevista com os pais ou cuidadores e professores, e avalia o comportamento adaptativo em 4 domínios: comunicação, atividades de vida diária, socialização e habilidades motoras. Os resultados dos testes neuropsicológicos devem ser interpretados com cautela e alguns fatores devem ser levados em consideração, como o contexto étnico e cultural, o nível educacional, a motivação, a cooperação e as deficiências associadas a DI.
DIAGNÓSTICO DIFERENCIAL DA DEFICIÊNCIA INTELECTUAL26
Alguns transtornos podem afetar a capacidade cognitiva e o comportamento adaptativo. São condições que mimetizam a DI e estão associados com alguma outra disfunção, entre elas estão a depressão, os déficits auditivos, visuais, transtornos de aprendizado e algumas síndromes epilépticas. Outros transtornos estão associados à DI, como a paralisia cerebral e transtornos do espectro autista (TEA). Nos casos de paralisia cerebral, as habilidades motoras estão sempre mais comprometidas em relação às habilidades cognitivas, e nos TEA as habilidades sociais adaptativas e linguagem estão mais afetadas.
TRATAMENTO DA DEFICIÊNCIA INTELECTUAL23,25,27
A DI não tem um tratamento específico, porém as deficiências associadas são, algumas vezes, passíveis de intervenção e tratamento farmacológico. A DI pode estar associada a comportamentos desafiadores (agressão, transtorno opositor-desafiante), assim como enfermidades mentais como transtorno do humor, ansiedade, epilepsia e transtornos comportamentais. A utilização do medicamento vai depender da necessidade, como no caso do transtorno do déficit de atenção e hiperatividade (TDAH) com a utilização de psicoestimulantes, nos comportamentos autolesivos e agressivos com o uso de neurolépticos, na depressão e transtorno obsessivo-compulsivo com a utilização de inibidores da recaptação de serotonina. Ocorre com frequência a associação de DI e comportamento agressivo e, também, autoflagelação, estando presentes em síndromes com X frágil, Smith-Magenis, Rett, Prader-Willi. A intervenção farmacológica pode ser feita com inibidores da serotonina ou buspirona.
A criança com DI necessita de suporte médico com acompanhamento frequente do pediatra, da participação da família do paciente e da escola. A estratégia para o manejo da criança com DI abrange vários aspectos da sua vida como saúde, educação, atividades sociais e de lazer, tratamento das doenças associadas e os problemas de comportamento.
A DI geralmente necessita da participação de vários profissionais, além do pediatra. Entre os profissionais envolvidos estão o psicólogo, fisioterapeuta, nutricionista, assistente social, fonoaudiólogo, enfermeiros e terapeuta ocupacional, assim como especialistas da área médica como neuropediatra, psiquiatra, geneticista e outras especialidades.
Com relação à educação, é importante que os programas pedagógicos sejam relevantes para as necessidades da criança e adaptados às suas habilidade individuais.
As atividades de lazer devem ser consideradas para as crianças com DI. Estas, geralmente, não encontram problemas quando inseridas em brincadeiras de crianças com desenvolvimento típico, enquanto que os adolescentes encontram mais dificuldades nas interações sociais e atividades de lazer.
A participação no esporte deve ser incentivada mesmo que não seja no aspecto competitivo, mas auxilia em alguns outros pontos como perda de peso, desenvolvimento da coordenação motora, da capacidade cardiovascular e melhora da autoestima. As atividades sociais também são importantes, como passeios, participação nos eventos típicos, encontros, danças etc.
Com relação à família, muitas se adaptam ao filho com DI, porém, outras têm dificuldades emocionais ou sociais. Os riscos de depressão nos pais, assim como abuso e negligência dos mesmos com a criança são frequentes quando comparados com crianças típicas.
Alguns fatores influenciam a família quanto à aceitação do problema da criança e o manuseio com a mesma, entre eles a estabilidade no relacionamento do casal, número pequeno de irmãos, autoestima dos pais, nível socioeconômico mais elevado, participação de toda a família, assim como a participação de programas de apoio na escola e na comunidade.
A DI de origem genética apresenta risco de transmissão a outros membros da família, justificando a importância do estudo molecular para o cálculo de risco de recorrência para futuras proles e aconselhamento familiar.
PROGNÓSTICO DA DEFICIÊNCIA INTELECTUAL11
Em crianças com DI grave, o prognóstico fica evidente no início da infância. O desempenho dos indivíduos com DI depende da causa subjacente, do grau de comprometimento cognitivo e adaptativo, das patologias associadas, dos recursos da família e serviços de treinamento da escola e da comunidade que são oferecidos às crianças e à família.
A outra maneira de ajudar esses pacientes é por meio da prevenção, informando a população sobre o uso de álcool na gravidez, que é extremamente nocivo para o feto, gerando alterações intelectuais, físicas, adaptativas e irreversíveis.
CONCLUSÃO
Os elementos essenciais na avaliação da DI incluem uma ampla investigação na família, na história pré, peri e pós-natal do paciente, exame físico completo e exame neurológico, além dos testes laboratoriais que devem ser seletivos e, conforme o quadro clínico, sugestivo para elucidar a causa da DI desde o cariótipo convencional até o sequenciamento do exoma. Porém, este, por ter custo elevado para a grande maioria da população, ainda não é realizado de rotina.
O acompanhamento do paciente ao longo dos anos é necessário para o diagnóstico, com avaliação física e comportamental associada aos testes neuropsicológicos e ao aconselhamento genético que é de fundamental importância para vários casos de DI, auxiliando no tratamento e prognóstico.
As causas de DI podem ser genéticas ou ambientais e, apesar de técnicas moleculares modernas, quase 60% das causas de DI ainda não são identificadas. Porém, duas causas apresentam tratamento específico com a identificação precoce: no caso da fenilcetonúria, por meio do teste do pezinho, e a outra com medidas preventivas, alertando as mulheres grávidas a não consumirem álcool em nenhum período da gestação e impedindo o aparecimento da SAF com danos irreversíveis para o bebê, incluindo a DI.
REFERÊNCIAS
1. Vasconcelos MM. Mental retardation. J Pediatr. 2004; 80(Suppl 2):S71-S82.
2. American Association on Mental Retardation. Retardo Mental. Definição, classificação e sistemas de apoio. 2006; Diagnóstico: 50-95.
3. American Association on Mental Retardation. Retardo Mental. Definição, classificação e sistemas de apoio. 2006; Visão Geral e Desenvolvimento: 17-48.
4. Carvalho ENS, Maciel DMMA. Nova concepção de deficiência mental segundo a American Association on Mental Retardation-AAMR. 2003; 11(2):147-56.
5. Assumpção Jr FB. Retardo Mental. In: Rodrigues SD, Azoni CAS, Ciasca SM (eds.). Transtornos do desenvolvimento: da identificação precoce às estratégias de intervenção. Ribeirão Preto, SP. 2014; 10:161-80.
6. Harris JC. New Terminology for Mental Retardation in DSM V and ICD 11. Curr Opin Psychiatry. 2013; 26(3):260-2.
7. American Association on Intellectual and Developmental Disabilities (AAIDD). Definition of intellectual disability. Disponível em: http://aaidd.org/intellectual-disability/definition#.WsiteNTwbcs. Acessado em: 1 de outubro de 2014.
8. Maris AF, Barbato IT, Trott A, Montano MAE. Familial Mental Retardation: a review and pratical classification. Ciência & Saúde Coletiva. 2013; 18(6):1717-29.
9. Nardes F, Araujo APQC, Ribeiro MG. Mental retardation in Duchenne muscular dystrophy. Rio de Janeiro: J Pediatr. 2012; 88(1):6-16.
10. Fusão EF, Vilanova, LCP. Transtornos do neurodesenvolvimento. In: Rodrigues MM, Vilanova LCP (eds.). Tratado de Neurologia Infantil. São Paulo: Atheneu 2017; 13:371-416.
11. Moeschler JB, Shevell M, Committee on Genetics. Comprehensive Evaluation of the child with Intellectual Disability or Global Developmental Delays. Pediatrics. 2014; 134(3):e903-e918.
12. Global Burden of Disease Study 2013. Global, regional and national incidence, prevalence and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries.1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. London: Lancet.
13. Cabarcas L, Espinosa E, Velasco H. Etiologia del retardo mental em la infancia: experiencia em dos centros de tercer nível. Biomedica. 2013; 33:402-10.
14. Torrado M del Valle. Evaluación etiológica del retardo mental de origen genético. Algoritmo diagnostico y nuevas técnicas moleculares. Arch Argent Pediatr. 2009; 107(3):225-46.
15. Cernach MCSP, Lamarck ZDN, Colovati MES, Galera MF. Infertilidade e esterilidade. In: Brunoni D, Perez ABA (eds.). Genética Médica. São Paulo: Editor Nestor Schor. 2013; 5:77-96.
16. Ramos MA, Christofolini DM, Antonialli GPM, Coprerski B, Brunoni D. Deficiência intelectual. In: Brunoni D, Perez ABA (eds.). Genética Médica São Paulo: Editor Nestor Schor. 2013; 16:397-423.
17. Curry CJ, Stevenson RE, Aughton D, Byrne J, Carey JC, Cassidy S, et al. Evaluation of mental retardation: recommendations of a Consensus Conference: American College of Medical Genetics. American Journal of Medical Genetics. 1997; 72(4):468-77.
18. Ferreira VKL, Ferreira GVD, Lima JMB, Cruz MS. Desempenho intelectual na exposição alcoólica fetal: relato de série de 10 casos. J Bras Psiquiatr. 2013; 62(3):234-9.
19. Diament A. Erros inatos do metabolismo e deficiência intelectual. Revista de Deficiência Intelectual. 2012 jan/jul; 2(2):1-20.
20. Kim CA, Albano LMJ, Bertola DR. Genética na prática clínica. Síndrome do cromossoma X-Frágil: a importância do diagnóstico precoce na prevenção da deficiência mental. 2010; 23:403-16.
21. Rocha NB. Busca de microrrearranjos no cromossoma X em meninos com deficiência intelectual. Tese de Mestrado. 2014; 1-74.
22. Storniolo LMA, Gimenes PVS, Costa AR, Melo DG. Aconselhamento genético de famílias de pacientes com deficiência intelectual da APAE de São Carlos, São Paulo, Brasil. Cad Saude Colet. 2011; 19(3):375-83.
23. Swaiman KF, Ashwal S, Ferriero DM, Shor NF. Principles and practice. Global developmental delay and mental retardation/intellectual disability. 5 ed. Pediatric Neurology. 2012; 43(7):554-76.
24. Polanczyk GV, Lamberte MTMR. Transtornos de aprendizagem e deficiência intelectual, psiquiatria da infância e da adolescencia. 2012; 20:230-42.
25. Junior FBA, Kuczynski E. Deficiência mental. Tratado de Psiquiatria da Infância e Adolescência. 2003; 24:247-64.
26. Tynan WD. Cognitive Deficits: Practice Essentials, overview, diagnosis. 2014 abr; 1-11.
27. Battaglia A, Carey JC. Diagnostic evaluation of developmental delay/mental retardation: an overview. Am J Med Gen. 2003; 117C(1):3-14.
28. Shevell MI, Majnemer A, Rosenbaum P, Abrahamowicz M. Etiologic yield of subspecialists evaluation of young children with global developmental delay. J Pediatr. 2000 mai; 136(5):593-8.
29. Kjos BO, Umansky R, Barkovich AJ. Brain MR imaging in children with developmental retardation of unknown cause: results in 76 cases. AJNR. Am J Neuroradiol. 1990 out; 11(5):1035-40.
30. Lourov IY, Vorsanova SG, Kurinnaia OS, Zelenova MA, Silvanovich AP, Yurov YB. Molecular Karyotyping by array CGH in a Russian cohort of children with intellectual disability, autism, epilepsy and congenital anomalies. Mol Cytogen. 2012; 5-46.
31. Riegel M, Barcellos N, Mergener R, de Souza KRS, Leite JCL, Gus R, et al. Molecular cytogenetic Evaluation of chromosomal microdeletions: the experience of a public hospital in Southern Brazil. Clin Biomed Res. 2014; 34(4):357-65.
32. Lay-Son Guillermo, Espinoza K, Vial C, Rivera JC, Guzmán ML, Repetto GM. Chromosomal microarrays testing in children with developmental disabilities and congenital anomalies. J Pediatr. 2015; 91:189-95.
33. Linhares ND, Svartman M, Valadares ER. Diagnóstico citogenético de pacientes com retardo mental idiopático. J Bras Patol Med Lab. 2012; 48(1):33-9.
34. Ligt J de, Willemsen MH, Van Bom BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012; 367:1921-9.
35. Prota JRM. Sequenciamento completo do exoma para investigação etiológica da deficiência intelectual inespecífica. Tese de conclusão de curso do programa de especialização em avaliação de tecnologias em saúde do Instituto de Avaliação de Tecnologia e Saúde. Universidade Federal do Rio Grande do Sul; 2015.
36. Monteiro FP, Kok F. Sequenciamento do exoma na investigação de deficiência intelectual. Revista de Deficiência Intelectual. 2014; 4(7):12-6.
37. Miotto EC, Lúcia MCS, Scaff M. Avaliação Neuropsicologica e Funções Cognitivas. Neuropsi Clin. 2012; 3-33.
UFPa, Mestre em Neurociencias, Hospital Ofir Loyola, Neuropediatra, Professora Ajunta II de Neurologia - Belém, PA, Brasil
Endereço para correspondência:
Regina Célia Beltrão Duarte
Hospital Ophir Loyola
Av. Gov Magalhães Barata, 992 - São Brás
Belém - PA, Brasil. CEP: 66060-281
E-mail: rcbduarte@yahoo.com.br / normatrabalhoacademico@gmail.com
Data de Submissão: 10/04/2018
Data de Aprovação: 25/08/2018