
A fibrose cística é uma desordem autossômica recessiva com envolvimento de vários órgãos1. Embora a doença pulmonar seja a principal responsável pela morbidade e mortalidade, algumas crianças apresentam envolvimento do trato gastrointestinal (TGI). As manifestações do TGI são decorrentes, sobretudo, do acometimento pancreático e hepatobiliar (5,7%) e estão relacionadas a impactos no crescimento ponderoestatural1. Desta forma, o diagnóstico precoce pode melhorar o prognóstico e a qualidade de vida destas crianças2.
Fortes evidências tem demonstrado os benefícios da inclusão do teste de triagem para fibrose cística no período neonatal, possibilitando intervenções imediatas que minimizem os efeitos da má absorção no ganho de peso e crescimento destas crianças2. Além disso, crianças submetidas ao teste de triagem parecem apresentar diminuição das complicações pulmonares2,3. Os benefícios a longo prazo após a identificação da FC em um estágio inicial da doença já estão bem estabelecidos, sobretudo, em relação à melhora nutricional, redução da morbidade e uma atenuação do impacto do tratamento para o paciente3,4.
RELATO DE CASO
Criança do sexo feminino, 2 anos e 7 meses, encaminhada ao especialista para investigação de hepatomegalia associada a alterações de enzimas hepáticas. Referia ainda evacuações amolecidas a líquidas desde o nascimento e, mais recentemente, queda de cabelo e aumento do volume abdominal. Ao exame, mostrava-se desnutrida, com pele ressecada, cabelos finos e quebradiços, descorada, abdômen globoso com fígado palpável a nível da cicatriz umbilical e baqueteamento digital. Na ausculta pulmonar, os sibilos eram audíveis sem estetoscópio, com retração subcostal e tiragens. Na história pregressa, relatava quadros de sibilância persistente desde 8 meses de idade, com várias idas ao pronto socorro. As crises de sibilância eram acompanhadas de tosse, dispneia e cianose periférica com necessidade de medicações de resgate e, por vezes, corticoide sistêmico.
Por orientação do pneumologista pediátrico, fazia uso de corticoide de longa duração desde os 16 meses de vida, com necessidade de doses progressivamente maiores. Mãe relatou 4 episódios de pneumonia desde o nascimento, sendo o último com 2 anos e 6 meses. O teste de triagem neonatal não se mostrava alterado.
Na investigação diagnóstica, apresentou aumento de enzimas hepáticas, sobretudo, às custas de TGP, hipoalbuminemia e ultrassonografia com hepatomegalia e aumento da ecogenicidade hepática (Figura 1). Além do comprometimento hepatobiliar, a criança apresentava sinais de insuficiência pancreática com controle glicêmicos alterados. Foram realizadas 3 dosagens de cloro no suor todas acima de 60 (a primeira com valor de 156). Diante do diagnóstico de fibrose cística foi iniciado tratamento enzimático e de suporte para o quadro de sibilância. A radiografia de tórax (Figura 2) demonstrou presença de espessamento peribrônquico e aumento de trama. A cultura do lavado brônquico mostrou colonização por Pseudomonas Aeruginosa sensível à ciprofloxacina. Durante a internação foi necessário uso de antibioticoterapia para tratamento de infecção pulmonar e medidas de suporte nutricional. Permaneceu internada por mais de 10 dias para controle dos sintomas, recebendo alta para acompanhamento ambulatorial.
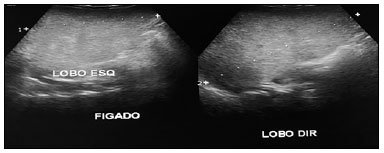 Figura 1. Ultrassonografia abdominal com aumento da ecogenicidade hepática.
Figura 1. Ultrassonografia abdominal com aumento da ecogenicidade hepática.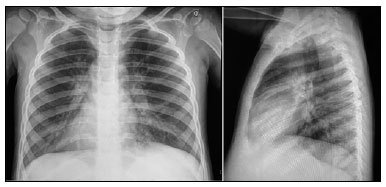 Figura 2. Radiografia tórax anteroposterior (AP) e perfil com alteração de trama.
Figura 2. Radiografia tórax anteroposterior (AP) e perfil com alteração de trama.Mais de 2.000 mutações estão associadas à FC. Estas diferenças moleculares, associadas a fatores individuais e relacionados ao meio ambiente resultam em fenótipos variados, dificultando o diagnóstico1. Atualmente, a maioria das crianças tem o diagnóstico da FC no período neonatal, ainda na fase assintomática, através do teste de triagem neonatal2,4,5. Estudo realizado em crianças americanas demonstrou que a triagem neonatal foi responsável pelo diagnóstico da FC em 62% das crianças6. Entretanto, alguns indivíduos são diagnosticados já na fase sintomática, talvez por não terem sido submetidos à triagem ou por terem apresentado um teste falso-negativo. Logo, em crianças que apresentam 1 ou mais sintomas de FC, deve se suspeitar da doença e serem monitoradas, independentemente da idade e do resultado do teste de triagem2,5.
O diagnóstico deve incluir a dosagem de cloro no suor e o teste genético para identificação das mutações associadas. As evidências demonstram benefício na identificação precoce pelo teste de triagem, com melhora do crescimento ponderoestatural2,5. Além disso, mostram diminuição das complicações pulmonares, embora os resultados sejam limitados em relação às mudanças no curso natural da doença. Por outro lado, crianças com diagnóstico tardio, que podem ter recebido um teste triagem falso-negativo, apresentam pior evolução da função pulmonar com infecções frequentes por Pseudomonas aeruginosa e aumento das internações hospitalares6. Estudos demonstram que anormalidades moleculares, celulares, teciduais e orgânicas acontecem desde os primeiros meses de vida e a progressão da doença tanto respiratória quanto do trato gastrointestinal foi demonstrada em crianças menores de 6 meses6. O comprometimento pulmonar é a forma clínica mais comum, podendo representar cerca de 92% dos casos6. Estudos sugerem que as alterações das vias aéreas na FC afetam a periferia pulmonar de forma mais extensivamente do que na asma.
A insuficiência pancreática é a segunda manifestação mais frequente e está presente em cerca de 90% das crianças, e se manifesta com uma síndrome de mal absorção com esteatorreia e comprometimento do ganho de peso7. O envolvimento hepático, embora não seja tão frequente, representa a terceira causa de morte. A hepatomegalia e a elevação das enzimas hepáticas pode ocorrer em mais de 20% dos pacientes. A esteatose hepática e colelitiase também podem ser observados e podem comprometer a função hepática6,7. No caso relatado, a criança apresentou um fenótipo com envolvimento pulmonar e pancreático, inclusive com comprometimento hepático.
O desenvolvimento de complicações em crianças menores incluindo infecções respiratórias com maior risco para colonização por Pseudomonas aeruginosa e efeitos deletérios irreversíveis na árvore pulmonar podem ocorrer por volta dos 2 anos de idade8. Muitos estudos demonstram que lactentes e pré-escolares representam um período crítico no qual mudanças estruturais no parênquima pulmonar pela FC podem iniciar8. O dano estrutural é decorrente de injúrias causadas pelos processos inflamatórios e infecciosos desde os primeiros anos de vida. Por outro lado, não há evidência que a identificação precoce destas alterações estruturais, como bronquiectasias, pode mudar o prognóstico destes pacientes8.
Em relação às manifestações digestivas, entre 59 a 71% das crianças estão fadadas a apresentarem insuficiência pancreática ao nascimento7. Naqueles que nascem com função pancreática normal, 16 a 20% terão comprometimento pancreático até por volta dos 6 meses1. Estudo realizado no Brasil em 55 crianças com diagnóstico de FC, a idade média do diagnóstico no grupo com doença hepatobiliar foi de 7 meses, bem mais precoce que no grupo sem envolvimento hepatobiliar, e ocorreu como manifestação clínica inicial em 55,6% dos pacientes9. No caso descrito, a criança mostrava-se sintomática desde os primeiros meses de vida, com episódios de diarreia e, posteriormente, sintomas pulmonares com piora progressiva a partir dos 8 meses de vida1,6. E, aos 2 anos de idade, quando do diagnóstico, já demonstrava doença pulmonar avançada com limitação da função expiratória e insuficiência pancreática com hiperglicemia, conforme relatado na literatura. Desta forma, o diagnóstico precoce e início do tratamento entre 4 e 13 meses de vida, estão associados a um melhor prognóstico da doença1,6.
CONCLUSÃO
Em conclusão, o fenótipo pulmonar é o mais comum, embora manifestações extrapulmonares também aconteçam e sejam clinicamente relevantes. Em crianças ocorre o comprometimento do crescimento pônderoestatural e da qualidade de vida, com aumento da necessidade de internações hospitalares. Os testes de triagem neonatal reduziram significativamente a média de idade do diagnóstico da FC e permitiram o tratamento precoce, reduzindo as internações e a morbimortalidade. Entretanto, o diagnóstico deve ser lembrado em todas as crianças sintomáticas com manifestações pulmonares e/ou extrapulmonares sugestivas, mesmo com teste de triagem neonatal normal.
O acesso precoce e rápido aos cuidados de saúde por equipe multidisciplinar permite intervenções imediatas em crianças menores com impactos na evolução da doença em idades maiores, embora seja insuficiente para prevenir o comprometimento anatômico e funcional do sistema respiratório ao longo do tempo.
REFERÊNCIAS
1. Santos ALM, Santos HM, Nogueira MB, Távora HTO, Cunha MLJP, Seixas RBPM, et al. Cystic fibrosis: clinical phenotypes in children and adolescents. Pediatr Gastroenterol Hepatol Nutr. 2018 Out;21(4):306-14.
2. Neemuchwala F, Taki M, Secord E, Nasr SZ. Newborn screening saves lives but cannot replace the need for clinical vigilance. Case Rep Pediatr. 2018 Jul;2018:7217326.
3. Course CW, Hanks R. Newborn screening for cystic fibrosis: Is there benefit for everyone. Paediatric Respir Rev. 2019 Ago;31:3-5.
4. Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N et al. Diagnosis of cystic fibrosis: consensus guidelines from the cystic fibrosis foundation. J Pediatr. 2017 Fev;181(Supl 1):S4-15.e1.
5. Rosenfeld M, Sontag MK, Ren CL. Cystic fibrosis diagnosis and newborn screening. Pediatr Clin North Am. 2016 Ago;63(4):599-615.
6. Coffey MJ, Whitaker V, Gentin N, Junek R, Shalhoub C, Nightingale S et al. Differences in outcomes between early and late diagnosis of cystic fibrosis in the newborn screening era. J Pediatr. 2016 Fev;181:137-45.e1.
7. Van Devanter DR, Kahle JS, O’Sulliva AK, Sikirica S, Hodgkins PS. Cystic fibrosis in young children: a review of disease manifestation, progression, and response to early treatment. J Cyst Fibros. 2016 Mar;15(2):147-57.
8. Ranganathan SC, Hall GL, Sly PD, Stick SM, Douglas TA. Early lung disease in infants and preschool children with cystic fibrosis. what have we learned and what should we do about it? Am J Respir Crit Care Med. 2017 Jun;195(12):1567-75.
9. Nascimento FS, Sena NA, Ferreira TA, Marques CDF, Silva LR, Souza EL. Hepatobiliary disease in children and adolescentes with cystic fibrosis. J Pediatr (Rio J). 2018 Set/Out;94(5):504-10.
Universidade Federal de Uberlândia, Pediatria - Uberlândia - Minas Gerais - Brasil
Endereço para correspondência:
Priscila Camargos Dumont Costa
Universidade Federal de Uberlândia
Av. João Naves de Ávila, nº 2121, Santa Mônica
Uberlândia, MG, Brasil. CEP: 38408-100
E-mail: priscilacdcosta@gmail.com
Data de Recebimento: 01/10/2019
Data de Aprovação: 13/12/2019
