
A malformação de Abernethy é uma malformação vascular congênita do território esplâncnico resultante da persistência de vasos embrionários, caracterizada pela existência de um shunt portossistêmico extra-hepático.1,2,3,4 É classificada em duas categorias de acordo com a anatomia.5 No tipo I , o mais frequente, há drenagem completa da circulação portal, de forma independente - tipo Ia - ou em um tronco comum - tipo Ib - na veia cava inferior.5 É mais frequente no sexo feminino e frequentemente está associada à presença de tumores benignos e malignos, bem como outras malformações, principalmente cardíacas. A malformação do tipo II, mais frequente no sexo masculino, é caracterizada por circulação hepática hipoplásica com circulação colateral.1,6
A apresentação clínica é variável: a doença pode permanecer assintomática até à idade adulta, mas existem casos descritos em crianças com cerca de 1 ano de idade.2 Os sintomas podem envolver os sistemas hepático, neurológico, pulmonar e metabólico.4,5 Incluem os sintomas associados a shunt portossistêmico, tais como síndrome hepatopulmonar e encefalopatia hepática, e outros associados a malformações coexistentes, incluindo doença cardíaca ou sintomas secundários a lesão intra-hepática.1,2,5
A terapia não é amplamente definida, mas inclui a realização de um novo shunt portossistêmico, fechamento do pré-existente e transplante de fígado.1,2,7RELATO DE CASO:
Relatamos o caso de um menino de 19 meses com dados familiares irrelevantes e história pessoal de sibilos recorrentes.
Bom estado de saúde até os 14 meses de idade, quando apresentou doença exantemática viral com trombocitopenia (30.000 - 40.000 plaquetas/µL) e hepatoesplenomegalia associada. A plaquetopenia persistiu em avaliações analíticas associadas, mas a hepatoesplenomegalia desapareceu. Nesse período, ele esteve várias no pronto-socorro com diarreia prolongada. A triagem de intolerância ao glúten foi negativa e o teste de IgA foi normal.
Avaliado por cardiologista pediátrico, foi diagnosticada comunicação interauricular do tipo seio venoso, dilatação atrial e ventricular direitas e estenose pulmonar discreta no anel valvular/supravalvar. A Síndrome de NOONAN foi considerada e uma avaliação genética foi realizada, mas nenhum dimorfismo foi destacado. O teste de matriz (array) foi normal.
Dez dias antes da internação (19/12), iniciou quadro de tosse e congestão nasal, sem outros sintomas. No terceiro dia de doença, foi diagnosticado com bronquiolite não hipoxêmica e otite no pronto-socorro pediátrico, recebendo alta hospitalar com salbutamol e amoxicilina. Cerca de uma semana depois (28/12), apresentou piora clínica com falta de ar, prostração e recusa alimentar. À admissão apresentava-se pálido, com olhos fundos, polipnéico (Frequência respiratória ~ 30cpm), com tiragem torácica, sopro sistólico III/VI e estertores subcrepitantes à ausculta cardíaca e pulmonar. Apresentava também esplenomegalia (2-3cm abaixo do rebordo costal esquerdo) e fígado (3-4cm abaixo do rebordo costal direito), adenopatias cervicais e inguinais infracentimétricas bilaterais e pequenas lesões petequiais espalhadas pelos membros e face.
Os exames de sangue mostraram anemia hemolítica, trombocitopenia, leucocitose leve com predominância de neutrófilos e proteína C-reativa negativa (tabela 1). A radiografia de tórax estava normal. A ultrassonografia abdominal mostrou fígado homogêneo de tamanho normal, com veias hepáticas permeáveis, convergindo para veia cava inferior e arterialização da circulação hepática. Não foi possível identificar a veia porta. Não foram encontradas alterações morfoestruturais pancreáticas e o tamanho do baço estava no limite superior (eixo longitudinal de 8 cm), sem lesões focais. Outros exames para investigação posterior são apresentados na tabela 2.
Iniciou oxigenioterapia e corticoterapia oral em altas doses (prednisolona 4mg/kg/dia).
Durante a internação, houve melhora clínica com diminuição progressiva da oferta de oxigênio suplementar, desconforto respiratório e apirexia sustentada. Recebeu 6 dias de corticoterapia, quatro dos quais em alta dose.
No oitavo dia, o paciente apresentou piora clínica e laboratorial, com início de febre, piora do desconforto respiratório e prostração e, portanto, foi transferido para nossa Unidade de Terapia Intensiva Pediátrica (UTIP). A radiografia de tórax permaneceu normal. A ultrassonografia abdominal foi semelhante à anterior. Havia uma pequena quantidade de fluido puro na bolsa de Morrison.
Ecocardiograma com dinâmica septal sugestiva de hipertensão ventricular direita com dilatação das cavidades direitas e gradiente valvar pulmonar preservado (20mmHg).
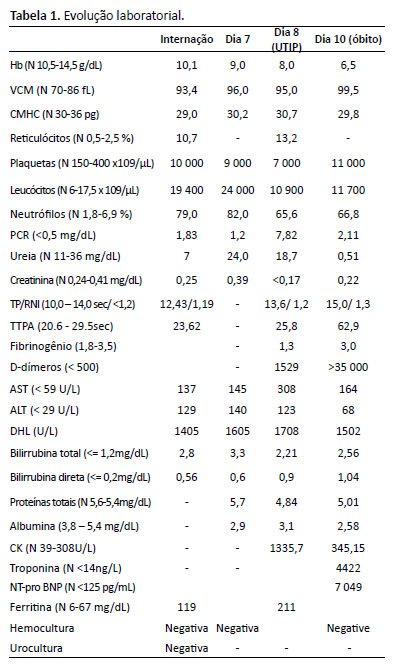
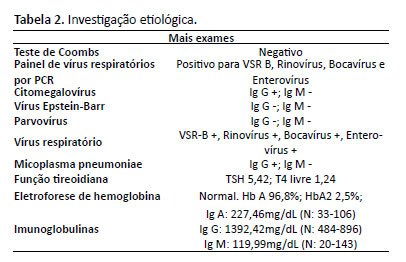
A angiotomografia computadorizada (angio-TC) de tórax e abdome mostrou dilatação aneurismática do tronco pulmonar (26 mm); pequena consolidação basal bilateral; coração aumentado, fígado homogêneo de tamanho normal; veias hepáticas patentes de tamanho normal convergindo para a veia cava inferior; sem veia porta no hilo, mas uma veia porta rudimentar com origem no hilo superior veia mesentérica em direção ao fígado direito; circulação arterial hepática proeminente; a veia esplênica e a veia mesentérica superior unem-se a um tronco comum que ascende como um shunt portossistêmico extra-hepático com varizes periaórticas, periesofágicas, perigástricas e esplenomegalia homogênea (Figuras 1 e 2). Não houve embolia pulmonar ou cavernoma portal.
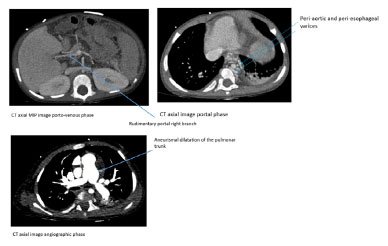 Figura 1. TC axial mostrando veia porta rudimentar (1/a) e sinais de hipertensão portal (2/b) e pulmonar (3/c).
Figura 1. TC axial mostrando veia porta rudimentar (1/a) e sinais de hipertensão portal (2/b) e pulmonar (3/c).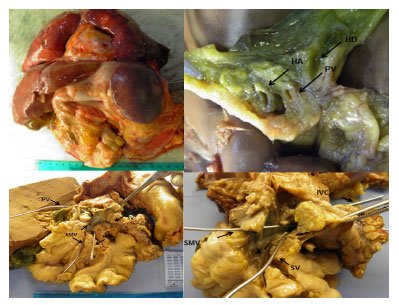 Figura 2. - Peças de necrópsia revelando hipoplasia da veia porta, vascularização pulmonar, malformações intra-abdominais e intra-hepáticas, alterações vasculares por hipertensão pulmonar grave (arteriopatia pulmonar plexogênica), cor pulmonale crônico, varizes esofágicas e atrofia do córtex cerebral. Legenda: BD: ducto biliar; HA: artéria hepática; IVC: veia cava inferior; PV: veia porta; SMA: artéria mesentérica superior; MSV: veia mesentérica superior; SV: veia esplênica.
Figura 2. - Peças de necrópsia revelando hipoplasia da veia porta, vascularização pulmonar, malformações intra-abdominais e intra-hepáticas, alterações vasculares por hipertensão pulmonar grave (arteriopatia pulmonar plexogênica), cor pulmonale crônico, varizes esofágicas e atrofia do córtex cerebral. Legenda: BD: ducto biliar; HA: artéria hepática; IVC: veia cava inferior; PV: veia porta; SMA: artéria mesentérica superior; MSV: veia mesentérica superior; SV: veia esplênica.Esses achados foram posteriormente interpretados como shunts portossistêmicos extra-hepáticos congênitos (CEPS) tipo 2, em que uma veia porta intra-hepática está intacta, mas parte do fluxo portal é desviado para uma veia sistêmica por meio de um shunt látero-lateral.
Considerando a gravidade clínica, apesar dos parâmetros inflamatórios negativos, iniciou antibioticoterapia com ceftriaxona (100mg/kg/dia) e azitromicina (10 mg/kg/dia).
Foi instituída terapia de suporte com transfusões (concentrados de hemácias e plaquetas) e oxigênio.
O quadro clínico continuou piorando com hipoxemia grave persistente, sem melhora com ventilação mecânica invasiva (ventilação convencional e oscilatória de alta frequência). Nesse momento havia disfunção sistólica bilateral e hipertensão pulmonar grave (~150mmHg) com dilatação importante das cavidades direitas e compressão das esquerdas ao ecocardiograma. Iniciou iloprosta endovenosa (2mcg/kg), nitroglicerina cutânea e óxido nítrico (40ppm), com discreta melhora.
Em poucas horas evoluiu com hipotensão refratária, piora da disfunção ventricular esquerda e progressão para falência de múltiplos órgãos. Após vários episódios de bradicardia extrema requerendo suporte avançado de vida, o paciente evoluiu para óbito.
Foi realizada necrópsia e encontrada hipoplasia da veia porta compatível com Malformação de Abernethy tipo II (fig. 2). Outros achados incluíram malformações vasculares pulmonares, intra-abdominais e intra-hepáticas, alterações vasculares por hipertensão pulmonar grave (arteriopatia pulmonar plexogênica), cor pulmonale crônico, varizes esofágicas e atrofia do córtex cerebral.
DISCUSSÃO
No presente caso, a doença se apresentou como uma síndrome hepatopulmonar, com hipertensão portal e pulmonar. No momento da admissão, a síndrome hepatopulmonar provavelmente foi desencadeada pelo episódio de infecção viral que culminou em hipertensão pulmonar grave, isquemia cardíaca e óbito. Retrospectivamente, os episódios prévios de dispneia e hipoxemia recorrentes provavelmente foram devidos, como relatado em outros casos5, à síndrome hepatopulmonar. Isso resulta da tríade: doença hepática, hipoxemia arterial e vasodilatação pulmonar.5 É importante ainda referir que a trombocitopenia persistente surge provavelmente no contexto de hipertensão portal, posteriormente confirmada pela presença de varizes esofágicas. Abernethy é uma das raras doenças em que podemos encontrar hipertensão portal sem cirrose hepática, o que atrasa o diagnóstico.
O diagnóstico é geralmente feito por imagiologia, principalmente a ecografia abdominal e angio-TC. No entanto, a angiografia digital é o padrão-ouro.5,7
O tratamento depende do tipo de malformação, manifestações clínicas e complicações existentes.5,7 Pode envolver fechamento de shunt portossistêmico, especialmente no tipo II, permitindo assim o desenvolvimento de circulação hipoplásica ou transplante de fígado.1,5 Embora o momento ideal para isso não esteja claramente definido, estudos mostram que mesmo na ausência de sintomas o tratamento precoce está associado à redução de complicações pulmonares e ótimo desenvolvimento psicomotor.5.6.7 No caso descrito, tratamentos prévios com fechamento da derivação sistêmica, segundo a literatura, levaram ao desenvolvimento da circulação hipoplásica e diminuíram as complicações da síndrome hepatopulmonar. Por outro lado, embora não existam sinais e sintomas de encefalopatia hepática, a autópsia descreveu sinais de atrofia cerebral provavelmente no contexto de doença do sistema nervoso central secundária a shunt hepatossistêmico.
CONCLUSÃO
As derivações hepatossistêmicas congênitas são raras, e a apresentação clínica é variável, portanto, o diagnóstico é um desafio. Elas podem se manifestar como sibilos recorrentes, uma entidade comum em crianças. Porém a CEPS deve ser considerada na presença de aspectos atípicos como a trombocitopenia deste caso. O diagnóstico precoce com reconhecimento oportuno da doença e terapia talvez pudesse ter evitado a evolução fulminante nesse paciente.
BIBLIOGRAFIA
1. Kang Z, Min X, Wang L. Abernethy Malformation Type II and Concurrent Nodular Hyperplasia in a Rare Female Case. Case Rep Radiol. 2018.
2. Ghuman SG, Gupta S, Buxi TBS, Rawat KS, Yadav A, Mehta N, et al. The Abernethy malformation - myriad imaging manifestations of a single entity. Indian J Radiol Imaging. 2016;26(3): 364–372.
3. Bernard O, Franchi-Abella S, Branchereau S, Pariente D, Gauthier F, Jacquemin E. Congenital portosystemic shunts in children: recognition, evaluation, and management. Semin Liver Dis. 2012; 32(4):2 73-87.
4. Kim ES, Lee KW, Choe YH. The Characteristics and Outcomes of Abernethy Syndrome in Korean Children: A Single Center Study. Pediatr Gastroenterol Hepatol Nutr. 2019;22(1):80–85.
5. Xie L, Li Y, Jiang J, Zhao J, Xiao T. A 10-year-old boy with dyspnea and hypoxia: Abernathy malformation masquerading as pulmonary arteriovenous fistula. BMC Pediatrics. 2019; 19(1), 1–5.
6. Azad S, Arya A, Sitaraman R, Garg A. Abernethy malformation: Our experience from a tertiary cardiac care center and review of literature. Ann Pediatr Cardiol. 2019;12(3):240-247.
7. Hu GH‚ Shen LG, Yang J‚ Mei JH‚ Zhu YF. Insight into congenital absence of the portal vein: Is it rare? World J, Gastroenterol 2008; 14(39): 5969-5979.
Hospital Prof. Doutor Fernando Fonseca, EPE, Pediatria - Amadora - Lisboa - Portugal
Endereço para correspondência:
Maria Maria Mendes.
Hospital Prof. Doutor Fernando Fonseca. IC19, 2720-276
Amadora, Portugal
E-mail: mariamariadamendes@gmail.com
Data de Recebimento: 28/08/2020
Data de Aprovação: 26/10/2020
