
Os quadros de crises convulsivas constituem manifestações clínicas frequentes das emergências pediátricas. O primeiro ano de vida de um lactente é crucial para o desenvolvimento neurológico, mediante sinaptogênese e mielinização crescentes e progressivas. Dessa forma, qualquer evento patogênico que ocorra nesse período de vida pode resultar em comprometimento cerebral significativo. Tais eventos podem muitas vezes iniciar de forma sutil e inespecífica quanto menor for a idade de apresentação, o que exige um elevado grau de suspeição clínica¹.
Dentre as causas metabólicas que desencadeiam episódios convulsivos no lactente, uma parcela de pacientes é portadora de erros inatos do metabolismo (EIM) intermediário que culminam em intoxicação aguda ou crônica. Essas condições clínicas são individualmente raras e se apresentam inicialmente com sintomas inespecíficos, o que gera atraso no diagnóstico e, consequentemente, no tratamento, podendo trazer danos irreparáveis ao sistema nervoso central da criança. Por outro lado, sabe-se que o diagnóstico e a intervenção precoce em pacientes com EIM podem resultar em evoluções clínicas favoráveis.
Atualmente, diversos testes de triagem metabólica incluem pesquisa de EIM, porém ainda não estão amplamente disponíveis para a população em geral. Portanto, deve-se incentivar que o esforço no reconhecimento inicial, diagnóstico precoce e manejo imediato seja difundido entre a comunidade de médicos que atuam na avaliação inicial de lactentes para melhor prognóstico dessas crianças2,3.
DESCRIÇÃO DO CASO
Em abril de 2023, um lactente do sexo masculino, com 6 semanas de vida, branco, deu entrada na emergência do Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG) no Rio de Janeiro, RJ, com relato de crise convulsiva tônico-clônica associada à cianose central e liberação esfincteriana, com duração de dez minutos, sem febre. A mãe da criança referia histórico de cinco crises de hipertonia com cianose iniciadas com 4 semanas de vida, de duração inferior a 1 minuto, revertidas espontaneamente, avaliadas em unidade de pronto atendimento próximo ao domicílio, sem prosseguimento de investigação etiológica até o momento.
História gestacional indicando mãe hipertensa durante segundo trimestre de gravidez, apresentou dez gestações prévias, com 7 filhos vivos de pai diferente e duas últimas gestações do mesmo pai, as quais culminaram em aborto espontâneo no primeiro trimestre. Não havia histórico de consanguinidade ou histórico de doenças metabólicas na família. Criança nascida de parto vaginal a termo, boa vitalidade ao nascer, sem intercorrências no período neonatal. A triagem metabólica neonatal não evidenciou alterações na avaliação de hemoglobinopatias, hipotireoidismo congênito, fenilcetonúria, hiperplasia adrenal congênita, deficiência de biotinidase, toxoplasmose congênita e fibrose cística.
À admissão na emergência, o lactente encontrava-se sonolento, afebril, pupilas isofotorreagentes, sem outras alterações. Exames laboratoriais incluíram: hemograma normal, glicemia 86 mg/dl, função hepática e renal normais, gasometria venosa com avaliação de lactato normal, ausência de distúrbios hidroeletrolíticos e elevação de amônia sérica (152 mmol/L; valor de referência < 30 mmol/L). A impressão inicial foi de crise convulsiva no lactente associada à hiperamonemia, sendo indicada internação hospitalar para investigação.
Durante internação, lactente realizou eletroencefalograma durante sono com padrão dentro da normalidade para faixa etária, ultrassonografia transfontanela e abdominal normais, avaliação de hormônios tireoidianos (TSH e T4L) normais, fundoscopia normal e exame do líquor com celularidade, bioquímica e lactato normais. Realizada avaliação pela Nutrologia por suspeita de etiologia metabólica das crises convulsivas, com indicação de realização de rastreio inicial para erro inato do metabolismo.
A investigação inicial incluía a avaliação laboratorial com rastreio metabólico (função hepática e renal, gasometria com avaliação de ânion gap, eletrólitos) normal, avaliação qualitativa de aminoacidúrias em amostra de urina congelada com resultado positivo e perfil TANDEM em papel filtro para análise quantitativa de aminoácidos e acilcarnitinas que evidenciou aumento discreto do aminoácido citrulina (resultado: 49,8 µmol/L; valor de referência 6-40 µmol/L).
No décimo dia de internação, apresentou episódio convulsivo tônico-clônico sem cianose, com duração de dois minutos, com exames laboratoriais no pós-ictal imediato indicando hiperamonemia mantida (93 mmol/L). Tais resultados aumentaram a suspeição etiológica de causa metabólica; realizado então ajuste dietético com redução de ingestão proteica (1,18 g/kg/dia), início do quelante de amônia benzoato de sódio 10% 100 mg/kg/dia e L-carnitina 10% 100 mg/kg/dia, e coletado painel genético com análise molecular por sequenciamento de nova geração para investigar variantes potencialmente patogênicas presentes em genes listados para distúrbios do ciclo da ureia e doenças tratáveis.
O resultado demonstrou presença da variante patogênica chr9:130,458,483 G>A, promovendo a substituição do aminoácido arginina na posição 86 por histidina (p.Arg86His) em heterozigose no gene ASS1 (argininosuccinate synthase 1), a qual já foi previamente associada ao quadro de citrulinemia tipo 1⁴.
Após diagnóstico molecular e controle das crises, o menor recebeu alta hospitalar para seguimento ambulatorial, com coleta seriada de exames laboratoriais. Manteve uso de benzoato de sódio 10% e L-carnitina, restrição proteica na dieta alimentar com uso de fórmula polimérica de partida fracionada em cinco vezes ao dia com três medidas para 100 mL de água filtrada, acrescidas de maltodextrina 5g e 3 mL de azeite em cada refeição, objetivando aporte de 115 kcal/kg/dia.
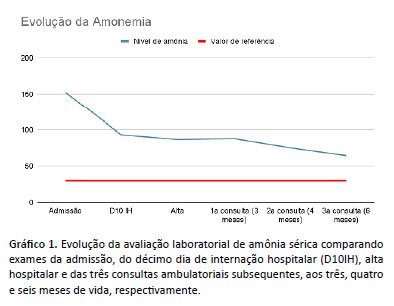
Evoluiu com manutenção da hiperamonemia em exames periódicos (Gráfico 1), sem crises convulsivas, sucedendo-se aumento gradual de benzoato de sódio 10% até 240 mg/kg/dia e L-carnitina até 150 mg/kg/dia, iniciado L-arginina 100 mg/kg/dia, associado à restrição proteica de 0,8-1 g/kg/dia. Aos seis meses de vida, houve melhora no controle do nível da amônia (62,3 µmol/L) e nova análise quantitativa por cromatografia de aminoácidos com resultado normal. A criança manteve desenvolvimento neuropsicomotor adequado segundo escala de Denver, ganho ponderal satisfatório e exame neurológico mantido normal para idade durante o seguimento clínico até os seis meses de idade.
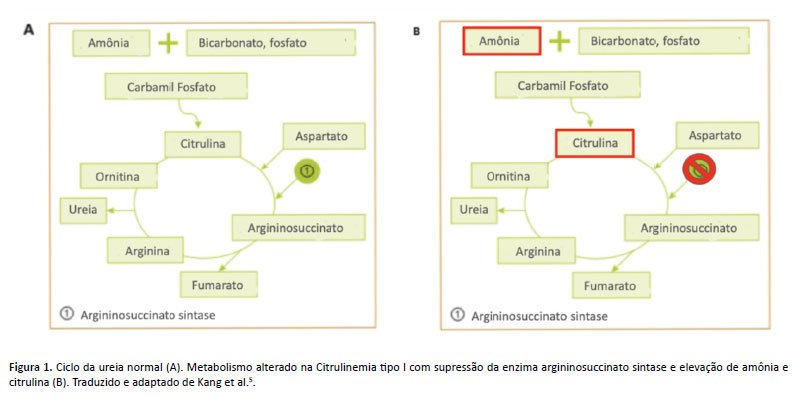
A citrulinemia tipo I (CTLN1) é um erro inato do metabolismo de herança autossômica recessiva causada pela mutação no gene ASS1, localizado no cromossomo 9q34.1, com a prevalência de 1 a cada 57.000 nascidos vivos, sendo a segunda doença do ciclo da ureia mais comum⁴. Ela é resultante da deficiência da terceira enzima do ciclo, a argininosuccinato sintase, distribuída em diversos tecidos, primordialmente no fígado, a qual catalisa a formação do argininosuccinato a partir de citrulina e aspartato4,5.
O ciclo da ureia consiste em uma série de reações enzimáticas que culminam na conversão da amônia, um metabólito tóxico que é liberado durante o processo de catabolismo proteico, em ureia, a principal escória nitrogenada, a qual é hidrossolúvel, excretada na urina e pouco tóxica ao organismo⁶. Quando os níveis de argininosuccinato sintase estão deficientes, a citrulina e o aspartato não podem ser convertidos em argininosuccinato, o que resulta em hiperamonemia e hipercitrulinemia, acompanhadas de deficiência de arginina (Figura 1)⁵.
A amônia é sabidamente tóxica para o cérebro, especialmente em fases precoces no neurodesenvolvimento neonatal, e sua concentração plasmática varia de acordo com o déficit enzimático específico e sua atividade residual plasmática, a ingestão proteica pelo indivíduo e a taxa de catabolismo endógeno, que pode estar afetada em situações de estresse metabólico, tais quais infecções4,6.
A CTLN1 apresenta-se clinicamente com letargia crescente e progressiva, sonolência, vômitos, hipotonia, convulsões e coma por hiperamonemia4,5. O diagnóstico pode ser realizado através de testes de triagem neonatal com análise quantitativa de citrulina em papel filtro. Em indivíduos sintomáticos sem testes de triagem, o diagnóstico é estabelecido através de hiperamonemia, aumento de citrulina plasmática e avaliação de alelos patogênicos no gene ASS1 em testes moleculares genéticos⁴. A avaliação e acompanhamento após o diagnóstico inclui consultas com nutrólogos, neurologistas e nutricionistas, com a avaliação seriada de níveis plasmáticos de amônia, gasometria, aminoácidos plasmáticos e eletrólitos.
O tratamento nutricional e medicamentoso envolve manutenção de baixos níveis de amônia plasmática através da restrição de proteína na dieta alimentar, de acordo com idade e sexo, e suas respectivas necessidades metabólicas. Deve-se monitorar o nível sérico de amônia e utilizar quelantes de nitrogênio, como benzoato de sódio, além da suplementação de arginina e L-carnitina4,5.
O prognóstico clínico e a possibilidade de sequelas neurológicas parecem ser relacionados ao nível de hiperamonemia durante o primeiro episódio sintomático, com 64% dos lactentes com níveis séricos < 180 mmol/L apresentando neurodesenvolvimento típico e apenas 8% em casos de amônia plasmática > 360 mmol/L4,7. Os riscos de déficits no neurodesenvolvimento, então, podem ser determinados a partir do nível plasmático de amônia e da duração do episódio de encefalopatia.
Portanto, verifica-se que a citrulinemia tipo 1 é um erro inato do metabolismo intermediário com quadro clínico amplo e muitas vezes inespecífico, principalmente em lactentes, podendo cursar com sintomas inicialmente leves, como hipotonia e sonolência, até repercussões agravantes como convulsões e coma. Deve haver alto grau de suspeição clínica para eventos sintomáticos sutis quanto mais tenra a idade e imatura a sinaptogênese do sistema nervoso central.
O prognóstico variável a curto e longo prazo é relacionado ao nível de hiperamonemia e à duração da encefalopatia. Logo, por ser uma doença tempo-sensível, torna-se imperativo o diagnóstico precoce, preferencialmente antes do desenvolvimento de sintomas, como em testes de triagem neonatal. Evidencia-se, portanto, a necessidade da suspeição clínica de EIM em quadros neurológicos agudos no lactente para objetivar um diagnóstico oportuno e o tratamento especializado com o intuito de atingir um desenvolvimento neuropsicomotor dentro do esperado para a idade.
REFERÊNCIAS
1. Baker PR 2nd. Recognizing and Managing a Metabolic Crisis. Pediatr Clin North Am. 2023;70(5):979-93. DOI: 10.1016/j.pcl.2023.05.009
2. Pavone P, Corsello G, Ruggieri M, Marino S, Marino S, Falsaperla R. Benign and severe early-life seizures: a round in the first year of life. Ital J Pediatr. 2018;44(1):54. DOI: 10.1186/s13052-018-0491-z
3. Leonard JV. Urea Cycle Disorders. In: Fernandes J, Saudubray JM, Van den Berghe G, Tada K, Buist NRM, eds. Inborn Metabolic Diseases. Berlin: Springer; 1995. p. 167-76.
4. Quinonez SC, Lee KN. Citrullinemia Type I. 2004 Jul 7 [Updated 2022 Aug 18]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington; 1993-2024. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK1458/
5. Kang H, Kim M, Lee JH. Nutritional Management in a Patient with Citrullinemia Type 1. Clin Nutr Res. 2021;10(3):268-77. DOI: 10.7762/cnr.2021.10.3.268
6. Rocha JC, Sequeira S, Cabral A, Almeida MF. Consenso para o tratamento nutricional das Doenças do Ciclo da Ureia. Acta Pediatr Port. 2009;40(2):83-93. Disponível em: https://www.spdm.org.pt/media/1117/acta-pediatr-port-2009_40_83_consenso_tratamento_doen%C3%A7as_ciclo_ureia.pdf
7. Ziogas IA, Wu WK, Matsuoka LK, Pai AK, Hafberg ET, Gillis LA, et al. Liver Transplantation in Children with Urea Cycle Disorders: The Importance of Minimizing Waiting Time. Liver Transpl. 2021;27(12):1799-810. DOI: 10.1002/lt.26186
Universidade Federal do Rio de Janeiro, Instituto de Puericultura e Pediatria Martagão Gesteira - Rio de Janeiro - Rio de Janeiro - Brasil
Endereço para correspondência:
Ricardo Mannato
Instituto de Puericultura e Pediatria Martagão Gesteira - IPPMG.
Rua Bruno Lobo, 50, Cidade Universitária
Rio de Janeiro, RJ, Brasil. CEP: 21941-912.
E-mail: ricardomannato@gmail.com
Data de Recebimento: 08/04/2024
Data de Aprovação: 06/05/2024
