
A epilepsia é um distúrbio cerebral complexo que envolve descargas elétricas cerebrais anormais, excessivas e sincrônicas dos neurônios de forma recorrente¹. Pode durar de segundos a minutos, afetando uma parte do cérebro e ficando restrita a essa região ou espalhando-se pelas demais áreas cerebrais². Para o diagnóstico, a International League Against Epilepsy (ILAE) dividiu em níveis, os quais consideram os recursos investigativos acerca da fisiopatologia específica que se desenvolve no paciente, terminando na investigação das possíveis etiologias, sem deixar de levar em conta as comorbidades.
São três níveis diferentes de investigação que conforme avançam ganham em detalhes e necessitam de mais recursos. Diante disso, sempre que possível, o diagnóstico deve ser estabelecido nos três níveis diferentes. No que diz respeito ao primeiro nível, o médico deverá confirmar que a crise é epiléptica, descartando os eventos paroxísticos não epilépticos.
No segundo nível, devemos classificar o tipo de epilepsia. Essa pode ser considerada generalizada, focal, generalizada e focal ou desconhecida. Uma crise que se inicia em uma região que abrange uma rede e que se estende de maneira bilateral é caracterizada como generalizada. Já as crises que, por meio do eletroencefalograma (EEG) ou da manifestação clínica, acontecem apenas em um hemisfério cerebral, geralmente restritas a uma rede subcortical, são descritas como focais. Um quadro de epilepsia também pode ser caracterizado como generalizado e focal, desde que apresente as duas formas no seu curso. Essa caracterização é feita por meio da investigação clínica, juntamente com os achados do eletroencefalograma.
Por fim, no terceiro nível temos o diagnóstico da síndrome epiléptica a partir dos achados clínicos, das manifestações no EEG e nos achados dos exames de imagem. Cabe ressaltar que algumas crises costumam se manifestar numa idade característica, possuem uma etiologia bem delimitada e, por consequência, também um prognóstico previsível. Além disso, muitas síndromes epilépticas são distintas o suficiente para serem identificadas por meio da investigação nos multiníveis, tais como a síndrome de West e de Dravet e a epilepsia de ausências na infância³ (Tabela 1).
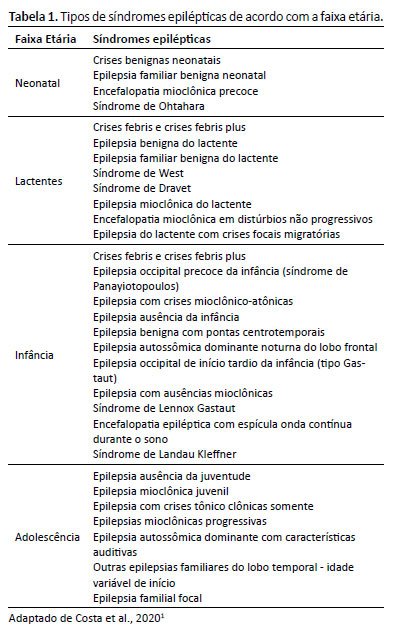
Ao se investigar a etiologia da epilepsia, depara-se com causas estruturais, genéticas, infecciosas, metabólicas, imunológicas ou desconhecidas. A etiologia infecciosa é a mais comum em todo o mundo. Origens estruturais são demonstradas em exames de imagem e podem ser secundárias a quadros infecciosos, traumatismos, acidente vascular cerebral, encefalopatia hipóxico-isquêmica ou genéticas, em função de malformações ou alterações em genes.
Quando a etiologia é genética, temos a comprovação por uma variante genética. Devemos lembrar que, apesar da origem genética, a presença de fatores desencadeantes epilépticos como a privação do sono, o estresse e outros não devem ser esquecidos. Para a etiologia metabólica, a epilepsia é secundária a um distúrbio conhecido ou presumido com manifestações clínicas sistêmicas. São exemplos de patologias a porfiria, a uremia, as aminoacidopatias ou as crises piridoxino-dependentes1-3.
No mundo, a prevalência da epilepsia varia de 10-15/1000 pessoas³. No Brasil, a estimativa é de 340 mil novos casos de epilepsia por ano, havendo 1,8 milhão de pacientes com epilepsia ativa¹. Já na faixa etária pediátrica e dos adolescentes chama atenção a incidência de crises em menores de 1 ano, estimada em 5/1000 nascidos vivos¹.
Diante do amplo espectro de epilepsias e da sua variada etiologia, além da alta prevalência na população geral e pediátrica, nota-se que o uso de ferramentas clínicas com vistas a direcionar a investigação e/ou facilitar o diagnóstico são necessárias. Assim, este trabalho teve por objetivo a revisão de casos de epilepsia em que foi aplicado um protocolo elaborado pelos autores, considerando os seguintes critérios: crises sem controle total, sem etiologia definida e sob avaliação genética.
MÉTODO
Estudo retrospectivo do tipo série de casos, constituída por crianças internadas em qualquer unidade pediátrica do Hospital Universitário ou acompanhadas no Ambulatório de Egressos da UTI Neonatal que realizaram avaliação genética, por apresentarem crises convulsivas sem etiologia definida. Um protocolo clínico foi elaborado para a reavaliação de crianças que apresentavam crises convulsivas pouco ou não responsivas aos anticonvulsivantes e com desenvolvimento atrasado (Anexo 1).
O protocolo foi adotado como rotina a partir de 2019 e continha variáveis como dados pré e perinatais, história familiar, tipo e evolução das crises, marcos do desenvolvimento, medicações utilizadas e exames realizados. Os dados foram coletados por um residente de pediatria que revisou os prontuários e as consultorias genéticas referentes ao período de 2019 a 2022 e selecionou aqueles em que o protocolo foi aplicado. Foram incluídos todos os pacientes que tinham de 0 a 10 anos e excluídos aqueles que não realizaram avaliação genética.
Todos os pacientes passaram por avaliação pediátrica, neurológica e genética. Além da avaliação genética clínica, foram colhidos painéis genéticos incluídos em programas de apoio diagnóstico, cedidos gratuitamente, dos Laboratórios Mendelics e Fleury. A interpretação do laudo dos painéis genéticos seguiu a classificação de variantes do American College of Medical Genetics (ACMG). Após o resultado dos painéis, foi realizado Aconselhamento Genético (AG), conforme recomendado, por uma geneticista local. De acordo com a variante encontrada, poderá haver reclassificação futura, sendo o acompanhamento genético importante para tal.
Os dados foram coletados respeitando o sigilo, não permitindo a identificação dos pacientes e tabelas foram analisados por meio do programa Microsoft Excel 2016 MSO versão (16.0). O presente projeto foi aprovado pelo Comitê de Ética em Pesquisa da Universidade Federal do Rio Grande (CEP-FURG), CAAE 64099922.2.0000.5324. Para a realização do painel genético, todos os responsáveis assinaram termo de Consentimento Livre e Esclarecido (TCLE) do laboratório e a todos foi dado AG no ambulatório local, quando o resultado foi recebido. O laudo foi entregue em mãos e não foi disponibilizado a terceiros.
RESULTADOS
Sobre o histórico gestacional dos pacientes selecionados, a comorbidade mais prevalente foi a doença hipertensiva da gestação (DHEG), ademais, houve um caso de consanguinidade parental em terceiro grau. Nenhum apresentou escore APGAR menor do que sete no primeiro e no quinto minuto. Dos pacientes selecionados, três eram prematuros. Todos iniciaram as crises epilépticas antes dos 2 anos de idade. Outras características encontram-se na Tabela 2. Dois pacientes eram sindrômicos, ou seja, apresentavam malformações anatômicas, sendo que em um deles havia alteração cardíaca, que consistia na persistência do canal arterial, presença de forame oval patente e de estenose pulmonar, e o outro caso micrognatia.
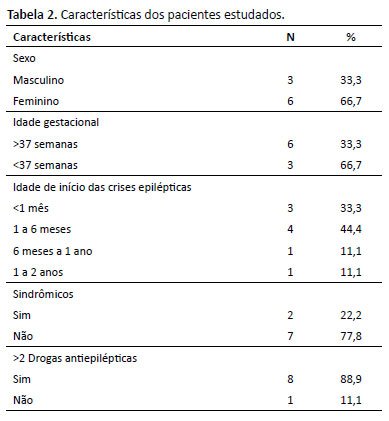
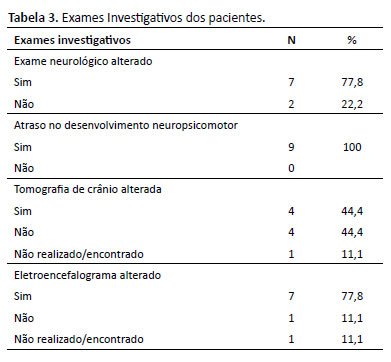
Ao exame neurológico, sete pacientes apresentaram hipotonia, clônus, hiperreflexia ou hiporreflexia, perda de força e alteração na marcha. Atraso no desenvolvimento neuropsicomotor (ADNPM), desde atraso na fala isolado ou em todos os domínios, foi apresentado por todos os pacientes. A tomografia e/ou ressonância do SNC estava alterada em quatro crianças, embora uma não detivesse tais dados. As alterações encontradas na ressonância nuclear magnética foram rarefação da mielina ou hipomielinização, focos de gliose, e alterações de sinal na sequência de difusão cortical envolvendo o hemisfério cerebral esquerdo, com predomínio nas regiões parietal, temporal e occipital, com áreas alteradas em corpo caloso e mesencéfalo. O EEG estava alterado em seis dos participantes, não havendo dados de um paciente (Tabela 3).
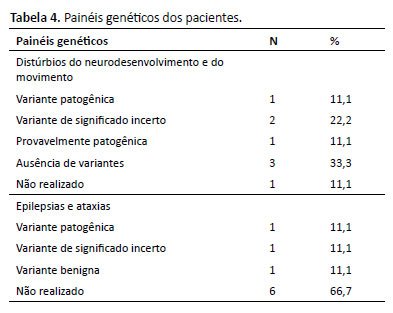
Nos painéis genéticos, primeiramente o de Distúrbios do Movimento e do Neurodesenvolvimento do Laboratório Mendelics, oito pacientes o realizaram, sendo que em um deles foi confirmada alteração gênica, um Erro Inato do Metabolismo (EIM), a Deficiência de Semialdeído Succínico Desidrogenase (SSADH), a ser detalhado adiante, um provavelmente apresenta alteração gênica, dois apresentam variante de significado incerto e dois não apresentaram alteração. Em um dos pacientes não foi realizado o painel, pois as crises epilépticas durante a investigação foram relacionadas a doença de base da criança, Cornélia de Lange.
O segundo painel genético, Epilepsias e Ataxias, apenas três pacientes o coletaram, já que o primeiro painel não havia mostrado alterações e o quadro era grave e persistente. Uma das crianças apresentou uma variante provavelmente patogênica em SCN2, cujo quadro clínico é muito compatível com a evolução (Tabela 4).
A partir da análise dos resultados, e de acordo com a literatura disponível sobre o tema, identificamos que o caso de EIM encontrado, mais precisamente a SSADH, é extremamente rara, com prevalência estimada de 1:460.000 casos nos países desenvolvidos4. Ao redor do mundo, aproximadamente 450 casos de SSADH foram identificados. A apresentação clínica conta com sintomas inespecíficos e a história natural da doença diferencia-se com a idade, mas durante a infância atraso global no neurodesenvolvimento, hipotonia, epilepsia e ataxia são frequentemente observados5. Ademais, os primeiros sintomas aparecem, em média, aos 11 meses de idade6.
Tais dados corroboram com aqueles encontrados no caso de EIM identificado nesse estudo. Fisiopatologicamente, essa doença leva ao acúmulo de ácido gama-hidroxibutírico, um metabólito neurotóxico, devido à não degradação do neurotransmissor inibitório GABA pela enzima semialdeído succínico desidrogenase. O diagnóstico dessa doença é de exclusão, sendo que o diagnóstico definitivo pode ser alcançado pela análise de fluidos corporais como a urina e o líquor, que mostrarão aumento do ácido gama-hidroxibutirico7.
Ademais, o diagnóstico também pode ser firmado através de investigação genética por sequenciamento de nova geração, quer seja por painel, caso do paciente investigado neste estudo, quer seja por sequenciamento de exoma. Em tais exames, se encontra uma mutação no gene ALDH5A1. No entanto, devido à apresentação clínica inespecífica que se sobrepõe a vários distúrbios neurológicos, o SSADH é provavelmente subdiagnosticado. Consequentemente, um alto nível de suspeita clínica é necessário para iniciar a investigação diagnóstica6.
Vale ressaltar que esse EIM é de herança autossômica recessiva, sendo necessário o aconselhamento genético8. As opções atuais de tratamento para a SSADH ainda são de suporte, mas existem contínuas tentativas de desenvolver terapias genéticas direcionadas4.
As variantes descritas nos resultados dos painéis genéticos são consideradas patogênicas quando impedem a produção de uma proteína normal, acarretando a alteração de alguma função essencial do organismo. As variantes provavelmente patogênicas são encontradas mais frequentemente em uma população afetada por uma patologia do que em uma população sadia, entretanto, essa variante ainda não foi documentada em bancos de variantes como patogênica, mesmo que a probabilidade para tal seja de 90%.
Existem, ainda, as variantes de significado incerto, conhecidas como VUS, cujo efeito no organismo é desconhecido. Quando o resultado é uma VUS nenhuma conduta deve ser tomada, embora seja recomendado que em algumas situações a variante seja reavaliada nos bancos clínicos se após um longo período o quadro persiste sem qualquer etiologia definida. A variante provavelmente benigna é aquela que não está associada a nenhuma doença em 90% dos casos, e a variante benigna é a que não está associada a nenhum quadro clínico9. Assim, os pacientes que receberam como resultado variante provavelmente patogênica estão propensos a serem diagnosticados com alguma condição genética. Tudo dependerá da correlação clínica e/ou da reclassificação da variante.
A epilepsia continua a ser uma doença marcada pela desigualdade. O subdiagnóstico e a falta de acesso ao tratamento são um dos problemas encontrados, principalmente, em países em desenvolvimento. No entanto, mesmo em países desenvolvidos, ainda existe uma desigualdade significativa no acesso ao diagnóstico preciso e tratamento específico para essa doença, que possui alta morbidade e mortalidade, especialmente no caso de pacientes com maiores complexidades10.
Diante do exposto, a necessidade de aprofundar a investigação em casos de epilepsia de difícil controle, acompanhadas por ADNPM, torna-se explícita. A criação e aplicação do protocolo clínico, avaliado por meio deste estudo, permitiu que a investigação se aprofundasse para os casos inconclusivos de epilepsia, o que se refletiu na definição diagnóstica e etiológica. Isso pode ser observado ao se analisar achados como o caso raro de um EIM e, também, na VPP em SCN2 cujo quadro clínico era muito compatível e continuou assim ao longo do tempo.
Dessa forma, a investigação permitiu constatar a razão genética como a causa em si e o AG das famílias, considerando uma doença autossômica recessiva e uma variante de novo, respectivamente. Além disso, o tratamento e as terapias foram encaminhados, conforme o diagnóstico. Tais resultados sugerem que a sistematização dos dados e a possibilidade de comparação auxiliam e agilizam uma investigação adequada e um melhor desfecho.
REFERÊNCIAS
1. Costa LLO, Brandão EC, Segundo LMBM, Atualização em epilepsia: revisão de literatura. Rev Med. 2020;99(2):170-81 [citado 2023 dez 2]. Disponível em: https://www.revistas.usp.br/revistadc/article/view/157412/160306
2. Nolasco MN, Ferreira WM, Rivero JRL. Epidemiologia dos casos de internação hospitalar por epilepsia no estado do Tocantins em 2018. Braz J Health Rev. 2020;3(6):17268-80 [citado 2023 dez 2]. Disponível em: https://www.brazilianjournals.com/index.php/BJHR/article/view/20729/16558
3. Souza LHL. Perfil epidemiológico da crise epilética da infância em um serviço de saúde terciário [Trabalho de conclusão de curso]. Passo Fundo: Universidade Federal da Fronteira Sul; 2021 [citado 2023 dez 2]. Disponível em: https://rd.uffs.edu.br/bitstream/prefix/4936/1/LUCAS%20H.%20LOPES%20DE%20SOUZA.pdf
4. Tokatly Latzer I, Bertoldi M, Blau N, DiBacco ML, Elsea SH, García-Cazorla À, et al. Consensus guidelines for the diagnosis and management of succinic semialdehyde dehydrogenase deficiency. Mol Genet Metab. 2024;142(1):108363 [citado 2024 mar 31]. Disponível em: https://doi.org/10.1016/j.ymgme.2024.108363
5. Phakey S, Rego T, Gaillard F, Panetta J, Evans A, De Jong G, et al. OCD symptoms in succinic semialdehyde dehydrogenase (SSADH) deficiency: a case report. BMC Psychiatry. 2020;20(1):395 [citado 2024 mar 31]. Disponível em: https://doi.org/10.1186/s12888-020-02794-8
6. Julia-Palacios NA, Kuseyri Hübschmann O, Olivella M, Pons R, Horvath G, Lücke T, et al. The continuously evolving phenotype of succinic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2024;47(3):447-62 [citado 2024 mar 31]. Disponível em: https://doi.org/10.1002/jimd.12723
7. Kirby T, Walters DC, Shi X, Turgeon C, Rinaldo P, Arning E, et al. Novel biomarkers and age-related metabolite correlations in plasma and dried blood spots from patients with succinic semialdehyde dehydrogenase deficiency. Orphanet J Rare Dis. 2020;15(1):261 [citado 2024 mar 31]. Disponível em: https://doi.org/10.1186/s13023-020-01522-5
8. Fattahi M, Bushehri A, Alavi A, Asghariazar V, Nozari A, Firouzabadi SG, et al. Bi-allelic Mutations in ALDH5A1 is associated with succinic semialdehyde dehydrogenase deficiency and severe intellectual disability. Gene. 2020:144918 [citado 2024 mar 31]. Disponível em: https://doi.org/10.1016/j.gene.2020.144918
9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24 [citado 2024 mar 31]. Disponível em: https://doi.org/10.1038/gim.2015.30
10. Pérez-Navarro VM, Cánovas-Iniesta M, Palazón-Cabanes B, Navarro-Lozano M. Epilepsia y desigualdad: descripción demográfica y análisis de la dificultad para el acceso a recursos avanzados en una población de un área de salud pequeña. Rev Neurol. 2023;77(11):259-65 [citado 2024 mar 31]. Disponível em: https://doi.org/10.33588/rn.7711.2023262
Faculdade de Medicina - Universidade Federal do Rio Grande, Pediatria - Rio Grande - Rio Grande do Sul - Brasil
Endereço para correspondência:
João Vitor Cândido Santos
Faculdade de Medicina - Universidade Federal do Rio Grande.
Rua Ramiro Barcelos, 2400, sala 416
Porto Alegre, RS, Brasil.
E-mail: joaovitorcandidosantos.jvcs@gmail.com

Data de Recebimento: 02/04/2024
Data de Aprovação: 16/05/2024
