
O CDC42, um produto do gene CDC42 — ciclo de divisão celular 42 —, é um membro da família de pequenas Rho GTPases, que desempenha um amplo espectro de funções fisiológicas na regulação do ciclo celular, incluindo adesão, polaridade, proliferação, migração, motilidade e ativação da transcrição celular1,2. A mutação do CDC42 resulta em um grupo heterogêneo de fenótipos e manifestações, uma vez que diferentes mutações missense já foram identificadas por toda a sequência de codificação desse gene pelo uso do sequenciamento completo de exoma3,4. A relação genótipo-fenótipo poderá ser melhor esclarecida à medida que mais pacientes forem identificados5.
As principais características clínicas dessa mutação incluem alterações no crescimento, dismorfismo facial, deficiência intelectual, defeitos cardíacos, anormalidades imunológicas, hematológicas e linfáticas e malformações cerebrais1,3. Dentro do espectro de manifestações, há a descrição de um novo fenótipo hematológico e autoinflamatório causado por uma mutação de novo no gene CDC42 em sua porção C-terminal, a síndrome NOCARH (Neonatal onset of pancytopenia, autoinflammation, rash and episodes of hemophagocytic lymphohistiocytosis), caracterizada por início neonatal de pancitopenia, autoinflamação, erupção cutânea e episódios de linfo-histiocitose hemofagocítica1,5. Até o presente momento, aproximadamente 15 casos dessa síndrome foram descritos em literatura6.
Para a identificação da mutação do gene CDC42 utiliza-se o sequenciamento completo do exoma3. O tratamento difere de acordo com as manifestações clínicas apresentadas pelos pacientes, dependentes do local da mutação. Por ser considerada como autoinflamatória, a síndrome NOCARH, em seus episódios agudos, como no caso relatado, pode ser manejada com diferentes classes de medicamentos, como corticoides, imunossupressores, imunomoduladores e anticorpos monoclonais. O tratamento definitivo consiste no transplante de células-tronco hematopoiéticas (TCTH)7.
Apresentamos neste relato de caso uma paciente de 17 anos com mutação do gene CDC42 relacionada a um quadro de resposta inflamatória sistêmica grave, diagnosticada com síndrome NOCARH e tratada com medicamento imunomodulador. Os termos de consentimento, tanto da paciente quanto da responsável legal, foram coletados, autorizando a publicação.
RELATO DE CASO
Paciente do sexo feminino, 17 anos, com história de nasofaringite aguda há 15 dias da internação, evoluindo com febre, sinais flogísticos em região cervical esquerda (dor, edema, hiperemia e calor local) e vômitos há 2 dias da admissão. Relata ainda perda ponderal de 6kg em 2 meses.
Internada, recebeu tratamento com ceftriaxona e clindamicina, realizada tomografia de tórax e região cervical, sendo identificado edema cervical anterior esquerdo e linfonodos reacionais, sem sinais de abscesso ou coleção. Evoluiu com choque séptico, insuficiência respiratória e necessidade de suporte em unidade de terapia intensiva. Foi entubada, iniciados fármacos vasoativos como vasopressina, noradrenalina, milrinona, corticoide endovenoso e vitamina C. Radiografia de tórax compatível com SDRA demonstrada na Figura 1. Modificado esquema de antibiótico para piperacilina-tazobactam, linezolida e fluconazol, sendo os antimicrobianos mantidos por 10 dias.
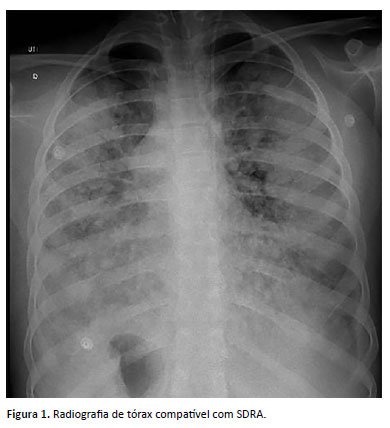
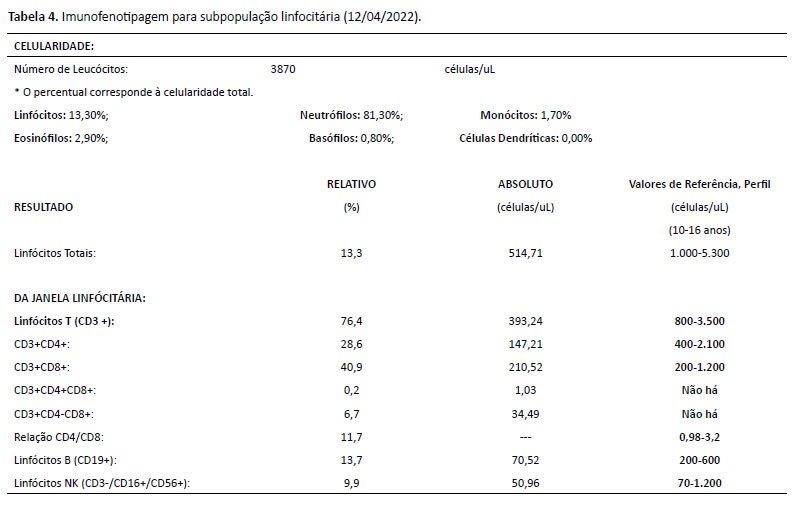
De história patológica pregressa, paciente sem intercorrências no período neonatal. Iniciou quadro de leucopenia e plaquetopenia crônicas aos 6 anos de idade, sendo iniciado seguimento conjunto com a hematologia. Investigada inicialmente para mielodisplasia, neoplasia e doenças reumatológicas, com resultados negativos. Conforme descritos nas Tabelas 1 a 4, temos os exames de investigação diagnóstica inicial (hemogramas mostrando a bicitopenia crônica; imunofenotipagem para população sublinfocitária, aspirado de medula óssea e dosagem de imunoglobulinas, com destaque para a imunoglobulina IgM, com valor acima da referência para a idade). Foi diagnosticada com a mutação do CDC42 através do exoma em setembro de 2019 (aos 13 anos), conforme exoma na Figura 2. Apresentou 2 internações hospitalares prévias (faringoamigdalite estreptocócica/ nasofaringite com plaquetopenia). Sem história familiar para imunodeficiência, avó com diagnóstico de lúpus eritematoso sistêmico.
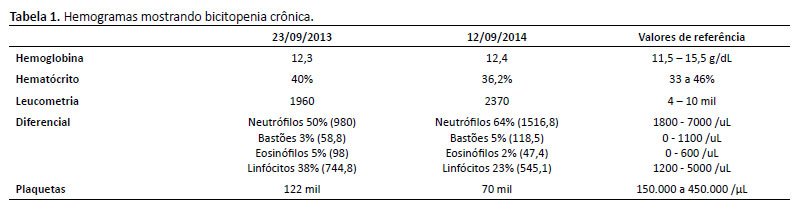
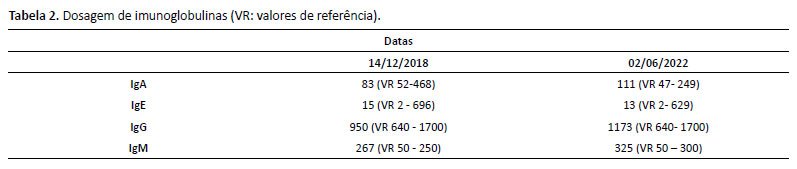
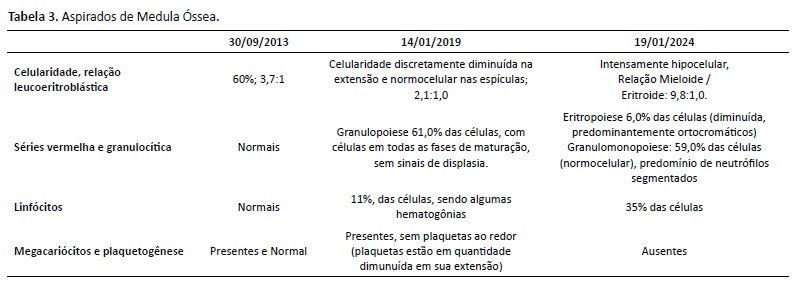

Após discussão em conjunto com as equipes da imunologia e reumatologia, foi aventada hipótese diagnóstica de Síndrome NOCARH. Nos exames laboratoriais, apresentava pancitopenia e desidrogenase lática elevada, além de outros achados sugestivos de síndrome hemofagocítica, concluindo clinicamente os critérios diagnósticos.
Indicada realização de tofacitinibe, com melhora importante após administração do medicamento. Atualmente em seguimento ambulatorial mensal com a equipe da reumatologia pediátrica, e, até a consulta de 3 meses após a alta hospitalar, sem novas intercorrências e sem efeitos adversos da medicação.
DISCUSSÃO
O diagnóstico e a delineação de novas síndromes genéticas são difíceis, dada a disponibilidade limitada de doentes, a heterogeneidade genética e variabilidade clínica1. Estudos recentes fornecem novas informações que expandem o espectro de doenças humanas associadas a mutações no CDC42. CDC42, um membro intracelular das GTPases da família Rho, é um regulador-chave da polaridade celular. Ao regular a montagem das estruturas do citoesqueleto de actina de maneira espaço-temporal, o CDC42, em parceria com o RAC e o RHO A, controla a forma e o movimento celular. Dessa forma, o CDC42 desempenha um papel importante na migração e nas funções dirigidas, processos que são essenciais para o desenvolvimento embrionário, hematopoiese, função do sistema imune, crescimento e a proliferação celular4.
Como GTPase, o CDC42 funciona como um interruptor molecular intracelular. Ele alterna entre duas formas — uma forma ativa quando ligada ao GTP, e uma forma inativa quando ligada ao GDP. Quando ativado, o CDC42 sofre uma alteração conformacional que lhe permite associar-se a membranas e interagir com moléculas efetoras, que por sua vez sofrem suas próprias alterações conformacionais para exercer funções bioquímicas a jusante4.
A partir de 2015, foram relatados os primeiros pacientes que apresentavam uma mutação missense heterozigótica de novo em CDC42 em p.Tyr64Cys, associada a uma constelação de características denominada síndrome de Takenouchi-Kosaki, que inclui dismorfismo, atraso no desenvolvimento, macrotrombocitopenia e, em um caso, imunodeficiência8,9. Em um estudo, Martinelli et al. (2018)10 relataram mais 15 pacientes com mutações missense heterozigóticas de novo no CDC42, vários dos quais eram recorrentes. As características da doença compartilhadas pelos pacientes incluíam crescimento deficiente, dismorfismo facial, anormalidades do desenvolvimento neurológico e malformações cardíacas, além de aberrações hematológicas e imunológicas10.
Mais recentemente, o uso mais amplo do sequenciamento completo do exoma em pacientes com condições complexas levou à descoberta de que fenótipos heterogêneos de doenças podem estar associados a mutações no mesmo gene. Lam et al. (2019)1 relataram pacientes com mutações missense heterozigóticas recorrentes de novo em CDC42 em p.Arg186Cys, uma variante não previamente associada à síndrome de Takenouchi-Kosaki. Esses pacientes apresentavam uma doença diferente, denominada NOCARH, caracterizada por início neonatal de pancitopenia, autoinflamação, erupção cutânea e episódios de linfo-histiocitose hemofagocítica (LHH)1.
A LHH foi caracterizada como uma síndrome clínica com hiperinflamação impulsionada pela ativação e expansão excessivas de macrófagos e linfócitos T CD8+. As características típicas incluem febre alta persistente, esplenomegalia, acometimento hepático, distúrbios da coagulação associada a pancitopenia e, geralmente, aumento da ferritina. Os mecanismos subjacentes a essa doença são diversos. Mutações que levam a uma citotoxicidade defeituosa por natural killer (NK) e linfócitos T CD8+ são a causa típica da LHH monogênica, tipicamente denominada LHH primária. No entanto, um número significativamente mais elevado de doentes apresenta LHH na ausência de citotoxicidade geneticamente defeituosa no contexto de infecções, doenças inflamatórias reumáticas e malignidade1.
Apresentamos no relato uma paciente de 17 anos com mutação do gene CDC42 previamente conhecida que, relacionada a um quadro de faringite aguda, evoluiu com um quadro de resposta inflamatória sistêmica com grave repercussão, fechando então critérios clínicos para síndrome NOCARH, sendo feito tratamento com tofacitinibe, com boa resposta terapêutica. O tofacitinibe, medicamento de uma nova classe terapêutica (inibidor seletivo de JAK1, JAK2 e JAK3), age dentro das células inibindo a atividade das enzimas JAK quinases, o que impede a produção de citocinas inflamatórias, resultando em diminuição da resposta inflamatória. O tofacitinibe é um imunossupressor muito utilizado em artrite reumatoide e artrite psoriásica, que é seletivo de JAK, e reduz a proliferação e ativação de linfócitos, e que tem efeitos adversos graves infrequentes, sendo o mais comum infecções graves (efeito adverso presente em medicações imunossupressoras em geral). Outros efeitos adversos relatados são hiperlipidemia e diarreia, sendo então importante o controle de lipidograma, hemograma e função hepática trimestralmente. Um fator limitante de seu uso é o alto custo, mas no caso relatado foi conseguida doação em tempo hábil para o tratamento. Associadamente a este, foi usada alta dose de hidrocortisona, devido ao quadro de choque refratário. Previamente ao tratamento com tofacitinibe, foi tentado tratamento com imunoglobulina, o qual não foi tolerado, pois após a primeira infusão da imunoglobulina a paciente apresentou TRALI (lesão pulmonar aguda associada à transfusão), sendo então suspenso o plano de tratamento com imunoglobulina.
O objetivo geral do tratamento é a supressão e o controle da hiperinflamação e dos níveis elevados de citocinas7. Dentre as citocinas inflamatórias envolvidas, a interleucina 18 (IL-18) têm sido um biomarcador para o diagnóstico de NOCARH e síndromes relacionadas com a desregulação da função CDC42. O provável papel da IL-18 seria estimular a produção de IFN-γ. Isso ajuda a explicar por que a maioria dos pacientes descritos na literatura apresentam melhora após o tratamento com antagonistas de IL-1 e/ou IFN-γ1. Além disso, a inibição farmacológica da via de sinalização JAK1/2 é uma opção de tratamento potente conhecida para várias condições autoinflamatórias induzidas por IFN, com ação em fibroblastos, e pode levar ao controle clínico da autoinflamação6. Esse último mecanismo de ação citado foi o utilizado como opção terapêutica com sucesso no caso relatado.
CONCLUSÃO
Neste documento, fora apresentado um relato de caso de uma paciente do sexo feminino com quadro prévio de bicitopenia crônica e suspeita de imunodeficiência, em que já havia conhecimento da mutação do CDC42 pelo exoma, mas quando realizado o exame, ainda não havia a confirmação da patogenicidade dessa variante. No internamento, paciente evoluiu com quadro grave e apresentou critérios diagnósticos de síndrome NOCARH, sendo instituído tratamento específico.
À medida que mais pacientes são identificados, torna-se possível esclarecer melhor as relações genótipo-fenótipo. Portanto, são necessários estudos mais detalhados de outras variantes para elucidar a gênese de doença imunológica/autoinflamatória e por que alguns pacientes com as mesmas variantes podem não se manifestar da mesma forma. Claramente, há muito mais a ser estudado sobre como o gene CDC42 funciona fisiologicamente em humanos e sobre como sua mutação se apresenta clinicamente.
REFERÊNCIAS
1. Lam MT, Coppola S, Krumbach OH, Prencipe G, Insalaco A, Cifaldi C, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. 2019;216(12):2778-99.
2. Szczawinska-Poplonyk A, Ploski R, Bernatowska E, Pac M. A novel CDC42 mutation in an 11-year old child manifesting as syndromic immunodeficiency, autoinflammation, hemophagocytic lymphohistiocytosis, and malignancy: a case report. Front Immunol. 2020;11:318.
3. Martinelli S, Krumbach OH, Pantaleoni F, Coppola S, Amin E, Pannone L, et al. Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am J Hum Genet. 2018;102(2):309-20.
4. Su HC, Orange JS. The growing spectrum of human diseases caused by inherited CDC42 mutations. J Clin Immunol. 2020;40(4):551-3.
5. He T, Huang Y, Ling J, Yang J. A new patient with NOCARH syndrome due to CDC42 defect. J Clin Immunol. 2020;40(4):571-5.
6. Kapp FG, Kretschmer S, Beckmann CC, Wäsch L, Molitor A, Carapito R, et al. C-terminal variants in CDC42 drive type I interferon-dependent autoinflammation in NOCARH syndrome reversible by ruxolitinib. Clin Immunol. 2023;256:109777.
7. Astigarraga I, Gonzalez-Granado LI, Allende LM, Alsina L. Haemophagocytic syndromes: The importance of early diagnosis and treatment. An Pediatr (Engl Ed). 2018;89(2):124.e1-8.
8. Takenouchi T, Kosaki R, Niizuma T, Hata K, Kosaki K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay. Am J Med Genet A. 2015;167A(11):2822–5.
9. Motokawa M, Watanabe S, Nakatomi A, Kondoh T, Matsumoto T, Morifuji K, et al. A hot-spot mutation in CDC42 (p.Tyr64Cys) and novel phenotypes in the third patient with Takenouchi-Kosaki syndrome. J Hum Genet. 2018;63(3):387–90.
10. Martinelli S, Krumbach OHF, Pantaleoni F, Coppola S, Amin E, Pannone L, et al. Functional Dysregulation of CDC42 Causes Diverse Developmental Phenotypes. Am J Hum Genet. 2018;102(2):309–20.
1. Hospital Pequeno Príncipe, Departamento de Terapia Intensiva Pediátrica - Curitiba - Paraná - Brasil
2. Hospital Pequeno Príncipe, Departamento de Pediatria - Curitiba - Paraná - Brasil
Endereço para correspondência:
Iassiminy Santos Merhi
Hospital Pequeno Príncipe, Departamento de Terapia Intensiva Pediátrica, Curitiba, Paraná, Brasil.
Rua Desembargador Motta, 1070, Água Verde
Curitiba, PR, Brasil. CEP: 80250-060.
E-mail: iassiminy@gmail.com
Data de Recebimento: 31/08/2024
Data de Aprovação: 03/01/2025
