
INTRODUÇÃO
Granulomatose com poliangiite (GPA), previamente conhecida como granulomatose de Wegener, é vasculite necrotizante de pequenos vasos, tipicamente associada com granulomas inflamatórios, glomerulonefrite, envolvimento do trato respiratório superior e inferior e presença de anticorpos anticitoplasma de neutrófilos (ANCA)1. É a principal vasculite associada ao ANCA, com incidência 0.4 a 6.4 casos para 1.000.000 crianças, em coortes internacionais, oriundas da Europa e Estados Unidos2.
Apesar de rara, uma coorte brasileira demonstrou que 12,4% dos adultos diagnosticados com GPA apresentavam comprometimento na faixa etária pediátrica3, evidenciando a importância da suspeição e reconhecimento da doença ainda na infância. Por vezes o diagnóstico é confundido inicialmente com doenças mais prevalentes (infecciosas, neoplásicas e otorrinolaringológicas), de modo que a confirmação pode se dar tardiamente4.
Objetiva-se relatar um caso de GPA em uma adolescente, diagnosticada inicialmente com fusariose. Para isso, foram utilizados critérios classificatórios estabelecidos por European League Against Rheumatism (EULAR)/ Paediatric Rheumatology International Trials Organisation (PRINTO)/ Paediatric Rheumatology European Society (PRES) em 20105. Este trabalho foi aprovado sob número de certificado de apresentação de apreciação ética (CAAE): 85703325.4.0000.0144.
RELATO DE CASO
Paciente do sexo feminino, previamente hígida, 13 anos de idade, admitida em unidade de terapia intensiva (UTI) pediátrica devido à insuficiência respiratória com necessidade de ventilação mecânica. Menor apresentava tosse hemoptoica, dispneia progressiva, febre intermitente e perda ponderal de início há 5 meses. Havia registro de internação em hospital externo há 2 meses com uso prévio de oxacilina, ceftriaxona e cefepime, além de investigação negativa para tuberculose (teste tuberculínico não reator, baciloscopia, teste rápido molecular – TRM-TB – e cultura para fungos negativos no lavado broncoalveolar - LBA). Paralelamente à prévia hospitalização, surgiu nódulo flogístico em região glútea à esquerda que fora biopsiado e tratado com amoxicilina-clavulanato, sem resposta.
À admissão, tomografia computadorizada (TC) de tórax demonstrou nódulos pulmonares difusos e adenomegalia mediastinal. Laboratorialmente havia aumento da velocidade de hemossedimentação (VHS) 75mm/h, proteína C-reativa (19mg/dL), leucograma 14.670 células/µL e anemia (hemoglobina 10mg/dL) normocítica e normocrômica, com provas de hemólise e teste de Coombs direto negativos. Considerando a possibilidade de doença fúngica, foram iniciados empiricamente afotericina B lipossomal, cefepime, levofloxacino e sulfametoxazol/trimetoprima. Sorologia anti-HIV resultou negativa, bem como hemoculturas para bactéria e fungos. Apesar destas negativas, mediante a suspeita de embolia séptica, ecocardiograma foi realizado, com resultado normal.
Após 10 dias, a paciente evoluiu com resolução da febre, decréscimo de provas de atividade inflamatória e tolerou extubação. Ao despertar, queixou-se de dor intensa em membro inferior esquerdo e evoluiu com epistaxe. TC de seios da face evidenciou pansinusopatia. Devido à suspeita de trombose venosa profunda (TVP), ultrassonografia (USG) Doppler de membro acometido não detectou TVP. Em decorrência de persistência da dor, foi solicitada eletroneuromiografia que identificou comprometimento neuropático do nervo ciático, sendo prescrita gabapentina. E, também, para complementação diagnóstica, realizada ressonância magnética de neuroeixo sem alterações.
Tão logo houve estabilidade clínica, novo LBA foi coletado, com resultado de segundo TRM-TB e pesquisa de galactomanana negativos. A pesquisa micológica direta visualizou leveduras hialinas e, posteriormente, recebemos resultado de cultura para fungos positiva para Fusarium spp. A avaliação imagenológica da região glútea foi indicativa de celulite infecciosa associada a miosite do glúteo máximo (Figura 1), também atribuídas à provável infecção fúngica.
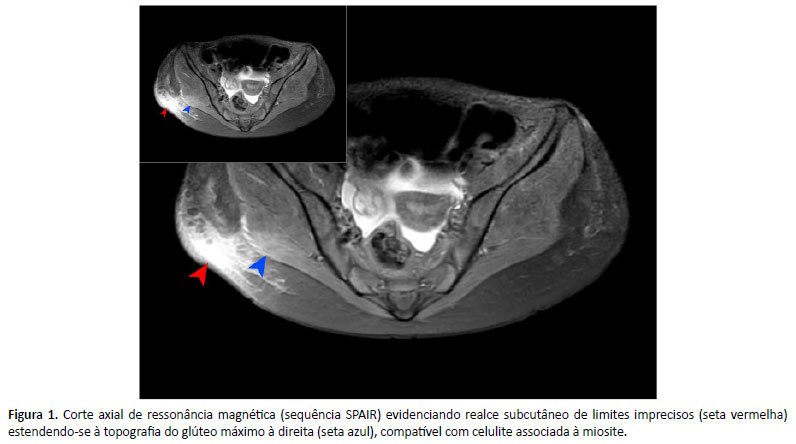
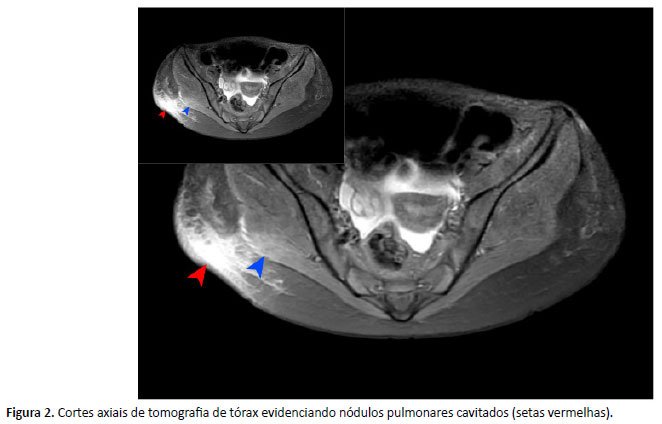
Cerca de 40 dias após admissão, a paciente recebeu alta com seguimento ambulatorial, em uso de voriconazol e gabapentina. Duas semanas depois, reiniciou tosse hemoptoica e febre, necessitando de reinternação, em que nova TC de tórax (Figura 2) evidenciou áreas de cavitação difusas bilateralmente e RM de órbitas com dacriocistite à direita, determinando discreta proptose, além de sinusopatia do esfenoide, maxilar e etmoidal. Outra broncoscopia foi realizada, em que se identificou espessamento ao nível da carina e mucosa friável.
Para diagnósticos alternativos, solicitaram avaliação da reumatologia após resultado de p-ANCA positivo 1:80. Além deste, houve positividade do Fator Antinuclear (FAN) em título de 1:320 padrão nuclear pontilhado fino, anticoagulante lúpico e antimieloperoxidase (MPO) reagentes. A pesquisa de autoanticorpos anti-DNA dupla hélice, c-ANCA, anti-Ro, anti-La, anti-Scl-70, anticardiolipinas e anti-B2-glicoproteína I resultou negativa. Para investigação de sarcoidose, a dosagem da enzima conversora da angiotensina (ECA) também evidenciou resultado normal.
Durante a reinternação, foi obtido laudo de biópsia cutânea de glúteo, indicando paniculite com infiltrado celular misto associada à necrose granulomatosa, que, em conjunto à avaliação reumatológica, corroborou o diagnóstico de vasculite associada ao ANCA. Para contemplar a avaliação renal, foram obtidas relação proteinúria/creatinúria (P/C) em amostra isolada de urina de 0.6 e função renal, que se mostrou preservada desde o início. No exame sumário de urina, não constavam elementos anormais e não havia alterações na sedimentoscopia.
Estabelecido o diagnóstico de GPA, a paciente recebeu pulsoterapia com metilprednisolona 30mg/kg por 3 dias consecutivos associada à infusão venosa de ciclofosfamida 750mg/m² de superfície corporal. Após 48 horas de corticoterapia, houve remissão da febre e decréscimo das provas de atividade inflamatória. A biópsia renal guiada por USG, realizada 2 semanas após pulsoterapia, com amostra de 29 glomérulos não mostrou alterações inflamatórias e apenas 1 esclerosado globalmente. Posteriormente ao procedimento, a paciente recebeu alta hospitalar em uso de prednisona 1.5mg/kg/dia, sulfametoxazol-trimetoprima, ácido acetil-salicílico, voriconazol, gabapentina, carbonato de cálcio e vitamina D.
DISCUSSÃO
Este caso demonstra a complexidade da jornada diagnóstica de uma adolescente que inicialmente apresentou febre, tosse hemoptoica e dispneia grave. Quando comparada à doença de início no adulto, a GPA pediátrica apresenta maior gravidade na primodescompensação, com maior necessidade de internação e suporte ventilatório4. Ademais, a doença na faixa etária pediátrica predomina em meninas, com média de início aos 14 anos6, ao passo que predomina em homens na maioridade2.
Inicialmente, a paciente foi tratada com diferentes betalactâmicos, sob hipótese de pneumonia bacteriana, entretanto sem melhora, e posteriormente, submetida à investigação de tuberculose pulmonar, doença endêmica no Brasil, com diagnóstico descartado. A boa evolução clínica, após terapia antifúngica, associada ao crescimento de Fusarium spp. em cultura de LBA, bem como relato de nódulo glúteo, preliminarmente, corroboraram o diagnóstico de fusariose. Os fungos do gênero Fusarium são ubíquos e amplamente distribuídos no ar, solo e água, sendo as formas mais comuns de infecção superficiais (onicomicose e ceratite). Em se tratando de imunocomprometidos, mais suscetíveis às formas invasivas, a doença se manifesta predominantemente por pneumonia (vidro fosco e nódulos), bronquiectasias e lesões de pele similares a ectima7.
A progressão das lesões pulmonares para cavitação e recrudescência dos sintomas iniciais motivaram a busca por diagnósticos diferenciais após a reinternação. Os resultados de p-ANCA e anti-MPO reagentes foram decisivos para o diagnóstico, pois, após sua obtenção, foram contemplados critérios classificatórios suficientes (3/5). O padrão de autoanticorpos evidenciados no caso em tela não é o mais característico da GPA (c-ANCA e antiproteinase 3), contudo destaca-se que ANCA reagente de qualquer tipo basta como critério e pode ser encontrado em até 90% dos casos5.
Destaca-se que, caso houvesse acesso precocemente ao laudo de biópsia cutânea, é possível que a suspeita clínica e a confirmação também ocorressem antecipadamente. Ao final, foram contemplados todos os 5 critérios classificatórios5 de GPA: histopatológico de pele, envolvimento de via aérea superior (epistaxe e sinusopatia), traqueobrônquico (estenose traqueal), pulmonar (nódulos cavitados), renal (relação P/C > 0.3mg/mg) e positividade do ANCA. A despeito de tantos critérios encontrados, ressalta-se que as alterações mais comuns são renais (83%), pulmonares (74%), e de vias aéreas superiores (70%)2.
A biópsia renal não corroborou o diagnóstico e uma possível justificativa foi a realização do procedimento posteriormente à pulsoterapia. O achado de glomerulonefrite pauci-imune e/ou necrotizante é consistente em 94% dos casos de GPA e é o maior determinante de morbimortalidade da doença6.
A maior coorte de GPA pediátrica demonstrou que a estenose subglótica é infrequente (10%), entretanto destaca-se em especificidade na doença de início na infância: cinco vezes maior quando comparada àquela em idade adulta. Sua inclusão nos critérios classificatórios EULAR/PRINTO/PRES reflete a relevância de tal lesão2.
Embora não sejam critérios para GPA, tanto a dacriocistite quanto mononeurite de ciático foram atribuídas à doença. Alterações oculares sob a forma de conjuntivite, episclerite, massas retro-orbitais e proptose podem ocorrer em até 40%, enquanto as alterações neurológicas mais graves como neuropatia, acidente vascular encefálico e fraqueza muscular ocorrem em menos de 3%6.
O tratamento proposto seguiu as recomendações para GPA com gravidade do SHARE (Single Hub and Access point for paediatric Rheumatology in Europe)8, inclusive no que diz respeito à terapia antiagregante profilática e sulfametoxazol-trimetoprima para profilaxia de Pneumocystis jirovecii. Devido à expectativa de imunossupressão prolongada, manteve-se profilaxia secundária para fusariose pulmonar.
CONCLUSÃO
O diagnóstico de vasculite primária em pacientes pediátricos é desafiador devido à sua ampla variedade de manifestações clínicas, que dependem da localização e extensão da doença, e à similaridade de apresentação de outras doenças. No caso em questão, as manifestações cutâneo-pulmonares inicialmente foram atribuídas em sua completude à infecção fúngica por Fusarium spp., porém o comprometimento de outros órgãos-alvo posteriormente, bem como o resultado de histopatológico e ANCA, foram cruciais para o diagnóstico. As vasculites devem ser consideradas como hipótese diagnóstica naqueles pacientes que se apresentem com inflamação sistêmica de origem inexplicada e/ou envolvimento multiorgânico.
REFERÊNCIAS
1. Bohm M, Gonzalez Fernandez MI, Ozen S, Pistorio A, Dolezalova P, Brogan P, et al. Clinical features of childhood granulomatosis with polyangiitis (Wegener’s granulomatosis). Pediatr Rheumatol Online J. 2014;12:18. DOI: https://doi.org/10.1186/1546-0096-12-18.
2. Cabral DA, Morishita K. Antineutrophil cytoplasmic antibody associated vasculitis. In: Petty RE, Laxer RM, Lindsley CB, Wedderburn LR, Mellins ED, Fuhlbrigge RC, editors. Textbook of paediatric rheumatology. 8th ed. Philadelphia: Elsevier; 2021:484-97. DOI: https://doi.org/10.1016/B978-0-323-24145-8.00036-3.
3. Belem JMFM, Pereira RMR, Perez MO, do Prado LL, Calich AL, Sachetto Z. Epidemiologic features of systemic vasculitides in the southeast region of Brazil: hospital-based survey. J Clin Rheumatol. 2020;26(7 Suppl 2):S106-10. DOI: https://doi.org/10.1097/RHU.0000000000001041.
4. James KE, Xiao R, Merkel PA, Weiss PF. Clinical course and outcomes of childhood-onset granulomatosis with polyangiitis. Clin Exp Rheumatol. 2017; [cited Year Month Day]; 35(Suppl 103):202-8. Available from: https://pubmed.ncbi.nlm.nih.gov/27749233/.
5. Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69(5):798-806. DOI: https://doi.org/10.1136/ard.2009.116657.
6. Cabral DA, Canter DL, Muscal E, Nanda K, Wahezi DM, Spalding SJ, et al. Comparing presenting clinical features in 48 children with microscopic polyangiitis to 183 children who have granulomatosis with polyangiitis (Wegener’s): an ARChiVe cohort study. Arthritis Rheumatol. 2016;68(10):2514-26. DOI: https://doi.org/10.1002/art.39729.
7. Nucci M, Anaissie E. Invasive fusariosis. Clin Microbiol Rev. 2023;36(4):e00159-22. DOI: https://doi.org/10.1128/cmr.00159-22.
8. de Graeff N, Groot N, Brogan P, Ozen S, Avcin T, Bader-Meunier B, et al. European consensus-based recommendations for the diagnosis and treatment of rare paediatric vasculitides: the SHARE initiative. Rheumatology (Oxford). 2019;58(4):656-71. DOI: https://doi.org/10.1093/rheumatology/key322.
Data de Recebimento: 02/05/2025
Data de Aprovação: 02/06/2025
Data de Publicação: 01/07/2026
