
INTRODUÇÃO
A imunodeficiência combinada grave (SCID - severe combined immunodeficiency) é uma condição caracterizada por defeito funcional e contagem significativamente baixa de células T, podendo haver, também, comprometimento de células B ou NK. A classificação fenotípica da doença baseia-se na presença ou ausência desses três tipos celulares1. É causada por mutações em genes que afetam o desenvolvimento do sistema imunológico, tornando os indivíduos extremamente vulneráveis às infecções oportunistas2.
Essa forma de imunodeficiência corresponde a 15% dos erros inatos da imunidade (EII), conforme registro da Sociedade Latino-Americana de Imunodeficiências (LASID)3. É considerada uma doença rara, estimada em 1 caso a cada 50.000 a 100.000 nascimentos2, embora este número seja subestimado pela mortalidade pré-diagnóstico, falta de diagnósticos nas manifestações clínicas atípicas e registros nacionais incompletos4. Sem intervenção adequada, as complicações infecciosas podem se agravar, tornando-se fatais até os 2 anos de idade3.
Apesar dos avanços na conscientização desta doença e nas tecnologias de diagnóstico e tratamento, a SCID ainda é um desafio em saúde pública no Brasil, devido ao atraso significativo no reconhecimento e tratamento das crianças afetadas3.
RELATO DE CASO
Lactente de 6 meses, masculino, nascido a termo e previamente hígido, com antecedente familiar de óbito do irmão aos 10 meses de vida por complicações de SCID. Apesar das orientações à família sobre o diagnóstico do filho falecido, o lactente deste relato não foi investigado ao nascimento ou encaminhado para seguimento com especialista. Realizou, entretanto, a triagem ampliada com a quantificação do número de círculos excisados de receptores de células T - TREC (T-Cell Receptor Excision Circles) no primeiro mês de vida, cujo resultado foi alterado (<10 cópias/µL), porém optou-se por repetir o exame, com resultado normal em recoleta (>10 cópias/µL). Assim, não se prosseguiu a investigação e recebeu as vacinas conforme o calendário do Ministério da Saúde, incluindo as de agentes vivos atenuados.
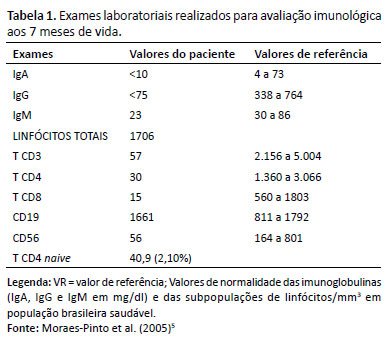
No sexto mês de vida, iniciou quadro respiratório, sendo diagnosticadas bronquiolite e pneumonia, e iniciado tratamento ambulatorial com amoxicilina. No nono dia da terapêutica, piorou clinicamente, somando dispneia e hipoxemia, sendo encaminhado para internação. A radiografia de tórax evidenciava infiltrado pulmonar difuso. Recebeu ceftriaxona e azitromicina, além de salbutamol e prednisolona, sem melhora do quadro. Por conseguinte, foi iniciada a avaliação imunológica (Tabela 1), que confirmou a hipótese de SCID, sendo iniciadas reposição de imunoglobulinas e profilaxias. A análise molecular, por sequenciamento de nova geração (NGS), evidenciou mutação no gene IL2RG, variante chrX:71.110.618C>A (p.Gly114Cys).
Pela ausência de melhora do quadro respiratório e imunodeficiência recém-identificada, foi ampliada a investigação para outras possíveis causas infecciosas. Material obtido de lavado gástrico revelou positividade para micobactéria. Introduzido, então, esquema com rifampicina, isoniazida, pirazinamida e etambutol. Não foi identificado DNA de citomegalovírus (CMV) em amostras de sangue; e pesquisa para vírus respiratórios, analisados por técnica de RT-PCR em material coletado por swab de naso-orofaringe, também foi negativa. A amamentação foi suspensa, pois a sorologia materna IgG para CMV era positiva.
Paciente evoluiu com melhora, mantendo-se estável até a transferência para realização do transplante de células-tronco hematopoiéticas (TCTH).
DISCUSSÃO
O caso clínico apresentado refere-se a uma doença rara, provavelmente subdiagnosticada, que representa uma emergência pediátrica. A idade e o estado de saúde ao diagnóstico/tratamento são fatores significativos no prognóstico. Estudos evidenciam que o tratamento definitivo com TCTH realizado antes dos 3,5 meses de idade resulta em 91.5% de sobrevida em 5 anos, comparado a 68% naqueles transplantados após esta idade6. O paciente relatado foi diagnosticado tardiamente, mas felizmente não apresentava graves complicações infecciosas e evoluiu favoravelmente após o transplante.
Como as crianças nascem assintomáticas e menos de um terço têm história familiar, a triagem neonatal para SCID tem extrema importância no diagnóstico e intervenção precoces, aumentando a sobrevida7. É realizada através da quantificação do número de TRECs, que são marcadores do desenvolvimento normal de linfócitos T produzidos pelo timo, obtidos do DNA da amostra de sangue coletada em papel-filtro especial, que pode ser realizada durante a coleta rotineira do teste do pezinho. Amostras com baixo número ou níveis indetectáveis de TRECs sugerem SCID, mas também podem identificar outras doenças que cursam com linfopenia de células T, como a Síndrome de DiGeorge8. Fatores como prematuridade, imunossupressores maternos ou infecção congênita também afetam o resultado. Diante de triagem alterada, testes confirmatórios são necessários e devem ser realizados idealmente antes da manifestação dos primeiros sintomas, o que não ocorreu com o paciente relatado. No Brasil, programas de triagem foram implantados recentemente em algumas cidades e estados, por iniciativas locais9. Há previsão, porém, de expansão do teste para todo o território nacional10, sendo necessária ampla divulgação da doença e criação de fluxos para atendimento após exames alterados.
Pacientes com SCID tendem a apresentar infecções recorrentes, geralmente persistentes, graves ou oportunistas, com frequência acometendo trato respiratório. Tosse crônica e sibilância devem levantar suspeita de pneumonia por pneumocystis jirovecii, citomegalovírus, micobactérias atípicas ou aspergillus4. A infecção pelo citomegalovírus, inclusive, aumenta o risco de doença enxerto-contra-hospedeiro (DECH) em pacientes que receberão transplante alogênico, sendo contraindicada a amamentação por mães soropositivas11. Quando há acometimento cutâneo, há maior susceptibilidade à invasão por estafilococos, estreptococos, enterococos, e bactérias Gram-negativas. Infecção por Candida e colonização por fungos na pele, orofaringe e intestino também são comuns4. Por vezes, bebês com SCID são vacinados com Bacillus Calmette-Guérin (BCG) antes do diagnóstico, e aproximadamente metade destes desenvolverão complicações relacionadas à vacina, sendo elas localizadas ou disseminadas, principalmente em pele, linfonodos ou pulmões12. Neste relato, o paciente havia recebido a vacina ao nascimento, que repercutiu com complicações pulmonares tardias, tratadas com esquema antituberculose. Assim, é essencial que o pediatra esteja atento aos sinais de alerta desde as primeiras consultas. Deve-se valorizar a história familiar e a pessoal de infecções recorrentes, dificuldade de ganho ponderal, internações por quadros virais exuberantes ou infecções incomuns para a idade. Candidíase oral persistente, dermatites extensas refratárias ao tratamento convencional ou reações graves a vacinas de vírus vivo também podem sugerir EII. A observação sensível e crítica desses detalhes clínicos pode ser decisiva para o diagnóstico precoce.
Os exames laboratoriais de rotina geralmente podem auxiliar no diagnóstico de SCID. Um hemograma com linfócitos <2.800 células/mm³ em lactentes nos primeiros meses de vida é altamente sugestivo da doença3. Embora uma contagem normal de linfócitos não descarte o diagnóstico, a presença de linfopenia em duas ocasiões indica a necessidade de fenotipagem de linfócitos e investigação dos diagnósticos diferenciais. Outro teste simples e útil é a radiografia de tórax, revelando ausência da sombra tímica, consistente com SCID. A medição de imunoglobulinas raramente auxilia no diagnóstico, pois concentrações normais de imunoglobulina G (IgG) no lactente jovem decorrem da passagem transplacentária, e níveis baixos de IgG na primeira infância podem ocorrer pela imaturidade imunológica. Números absolutos de subtipos linfocitários são mais úteis que porcentagens, e cada padrão fenotípico sugere um diagnóstico específico. Na SCID típica, teremos contagens de TCD3+ <300 células/ml ou <20% de TCD4 naive13. O paciente descrito preenchia os critérios da doença, e a complementação com a contagem de células B e NK definiu o subtipo T-B+NK-.
A avaliação genético-molecular objetiva identificar variantes genéticas ligadas ao defeito imunológico, sendo essencial para diagnóstico definitivo, condicionamento pré-transplante de medula óssea e aconselhamento familiar. Além das abordagens tradicionais, novas tecnologias, como o painel NGS, permitem detectar mutações com maior precisão3. No caso citado, o resultado NGS revelou mutação no gene IL2RG, ligado ao X, considerada a forma genética mais comum de SCID típica14, com incidência de 1:130.000 nascimentos4.
Atualmente, o TCTH é considerado o tratamento padrão-ouro e curativo para SCID. Em um cenário ideal, o paciente aguarda um doador HLA (antígeno leucocitário humano) compatível, seja aparentado ou não15. Até a realização do transplante, é fundamental que sejam adotadas medidas de cuidados pessoais, reposição de imunoglobulina humana, profilaxias, contraindicação de vacinas de patógenos atenuados e irradiação de hemoderivados que venham a ser necessários1,16. Apesar de o TCTH ser a opção mais eficaz, a presença de infecção ativa no momento do transplante reduz a sobrevida para 64,7%, frente a 82,3% nos que não apresentam infecção ativa5. Após transplante bem-sucedido, a maioria recupera a função imunológica, embora alguns ainda precisem de reposição ocasional de imunoglobulina1. No caso apresentado, o pai do paciente foi o doador haploidêntico, ocorrendo reconstituição imunológica após o transplante.
Além do tratamento convencional, a terapia gênica tem mostrado promissora evolução no tratamento da SCID. Envolve alterar geneticamente células-tronco hematopoiéticas do próprio paciente para corrigir as mutações responsáveis pela doença e reimplantá-las. No entanto, ainda se encontra em estágios iniciais de desenvolvimento, representando uma esperança para o futuro desses pacientes2,16.
CONCLUSÃO
Embora rara, essa condição precisa ser reconhecida precocemente pelos pediatras para evitar o surgimento de infecções potencialmente fatais. A triagem neonatal é essencial para a detecção precoce da doença, viabilizando o tratamento oportuno com o TCTH, considerado o padrão-ouro e curativo. O desafio atual consiste em estabelecer políticas públicas eficazes que ampliem a triagem para todo o território nacional, capacitem centros de referência e aumentem a disponibilidade do TCTH.
O relato do caso foi aprovado pelo Comitê de Ética em Pesquisa do ICEPi, sob o número de aprovação (CAAE) 85359424.0.0000.0375.
REFERÊNCIAS
1. Pfisterer JC, Martini SV, Errante PR, Frazão JB. Severe combined immunodeficiency: review of the literature. Braz J Allergy Immunol. 2014;2(2):56-65. DOI: https://doi.org/10.5935/2318-5015.20140010.
2. Cunha MLS, Nascimento JS, Salomão JPP, Martins RS, Freitas LO, Hernandes Junior PR, et al. Severe combined immunodeficiency (SCID): recent advances, ongoing challenges, and the need for global newborn screening. In: Lauer RD, editor. Ciências da saúde: bem-estar e qualidade de vida [Internet]. São Paulo: Editora Atena; 2023; [cited 2024 Aug 19]:211-6. DOI: https://doi.org/10.22533/at.ed.12523041219. Available from: https://atenaeditora.com.br/catalogo/ebook/ciencias-da-saude-bem-estar-e-qualidade-de-vida.
3. Aranda CS, Guimarães RR, Gouveia-Pereira Pimentel M. Combined immunodeficiencies. J Pediatr (Rio J). 2021;97(Suppl 1):S39-48. DOI: https://doi.org/10.1016/j.jped.2020.10.014.
4. Aranda CS, Gouveia-Pereira MP, Silva CJM, Rizzo MCFV, Ishizuka E, Oliveira EB, et al. Severe combined immunodeficiency diagnosis and genetic defects. Immunol Rev. 2024;322(1):138-47. DOI: https://doi.org/10.1111/imr.13310.
5. Moraes-Pinto MI, et al. Imunodeficiência primária: os 10 sinais de alerta [Internet]. 2005; [citado 2024 Ago 19]. Disponível em: http://www.imunopediatria.org.br/_download/10sinais.pdf.
6. Cuvelier GD, Logan BR, Prockop SE, Buckley RH, Kuo CY, Griffith LM, et al. Outcomes following treatment for ADA-deficient severe combined immunodeficiency: a report from the PIDTC. Blood. 2022;140(7):685-705. DOI: https://doi.org/10.1182/blood.2022016196.
7. Thakar MS, Logan BR, Puck JM, Dunn EA, Buckley RH, Cowan MJ, et al. Measuring the effect of newborn screening on survival after haematopoietic cell transplantation for severe combined immunodeficiency: a 36-year longitudinal study from the Primary Immune Deficiency Treatment Consortium. Lancet. 2023;402(10396):129-40. DOI: https://doi.org/10.1016/S0140-6736(23)00731-6.
8. Notarangelo LD. Genetically-determined defects of T cell development. Allergy Asthma Proc. 2024;45(5):326-31. DOI: https://doi.org/10.2500/aap.2024.45.240028.
9. Barreiros LA, Sousa JL, Geier CB, Piller AL, Kanegae MPP, França TT, et al. SCID and other inborn errors of immunity with low TRECs—the Brazilian experience. J Clin Immunol. 2022;42(6):1-22. DOI: https://doi.org/10.1007/s10875-022-01275-9.
10. Brasil. Lei nº 14.154, de 26 de maio de 2021. Diário Oficial da União [Internet]. 2021 maio 27; [acesso em 2024 Ago 19]. Disponível em: https://www.in.gov.br/web/dou/-/lei-n-14.154-de-26-de-maio-de-2021-322209993.
11. Kielsen K, Møller DL, Pedersen AE, Nielsen CH, Ifversen M, Ryder LP, et al. Cytomegalovirus infection is associated with thymic dysfunction and chronic graft-versus-host disease after pediatric hematopoietic stem cell transplantation. Clin Immunol. 2024;265:110302. DOI: https://doi.org/10.1016/j.clim.2024.110302.
12. Fekrvand S, Yazdani R, Olbrich P, Gennery A, Rosenzweig SD, Condino-Neto A, et al. Primary immunodeficiency diseases and Bacillus Calmette-Guérin (BCG)-vaccine-derived complications: a systematic review. J Allergy Clin Immunol Pract. 2020;8(4):1371-86. DOI: https://doi.org/10.1016/j.jaip.2020.01.038.
13. Dvorak CC, Haddad E, Heimall J, Dunn E, Buckley RH, Kohn DB, et al. The diagnosis of severe combined immunodeficiency (SCID): the Primary Immune Deficiency Treatment Consortium (PIDTC) 2022 definitions. J Allergy Clin Immunol. 2023;151(2):539-46. DOI: https://doi.org/10.1016/j.jaci.2022.10.022.
14. Hauer J, Poplack DG, Armsby C. Severe combined immunodeficiency (SCID): an overview [Internet]. Boston: UpToDate; 2022; [cited 2024 Aug 19]. Disponível em: https://www.uptodate.com/contents/severe-combined-immunodeficiency-scid-an-overview.
15. Goebel GA, de Assis CS, Cunha LAO, Minafra FG, Pinto JA. Survival after hematopoietic stem cell transplantation in severe combined immunodeficiency (SCID): a worldwide review of the prognostic variables. Clin Rev Allergy Immunol. 2024;66(2):192-209. DOI: https://doi.org/10.1007/s12016-024-08993-5.
16. Segundo GR, Condino Neto A. Treatment of patients with immunodeficiency: medication, gene therapy, and transplantation. J Pediatr (Rio J). 2021;97(Suppl 1):S17-23. DOI: https://doi.org/10.1016/j.jped.2020.10.005.
Data de Recebimento: 11/05/2025
Data de Aprovação: 29/09/2025
Data de Publicação: 01/07/2026
