INTRODUÇÃO
As mucopolissacaridoses (MPS) são um grupo de doenças de acúmulo de glicosaminoglicanos (GAGs) como dermatan sulfato, heparan sulfato, queratan sulfato e condroitina. Existem 8 tipos de MPS, enumeradas de I a IX, causadas por deficiência de uma das onze enzimas diferentes envolvidas no catabolismo dos GAGs1. Pacientes com MPS apresentam progressivo acúmulo de GAGs em diferentes tecidos, levando ao quadro clínico da doença.
Embora sejam doenças tipicamente de acometimento multissistêmico, algumas formas de MPS1, como a tipo IX, a mais rara de todas formas de MPS, não apresenta quadro neurológico, visceral ou cardiovascular, sendo apenas de acometimento osteoarticular, lembrando quadros de artrite reumatoide infantil. Já outras, como tipo III (síndrome de Sanfilippo), apresentam acometimento predominante do sistema nervoso central com menos achados somáticos quando comparada com outros subtipos como tipo I (síndrome de Hurler), tipo II (síndrome de Hunter) e tipo VI (síndrome de Marateaux-Lamy)1,2.
A mucopolissacaridose tipo III (MPS III), também conhecida como síndrome de Sanfilippo, é uma doença genética autossômica recessiva caracterizada pelo acúmulo de glicosaminoglicanos nas células lisossomais. É causada por deficiência de uma das quatro enzimas necessárias para a quebra e reciclagem do heparan sulfato1.
Há quatro subtipos comprovados em humanos de MPS III, que são tipo A (deficiência da heparan-N-sulfatase, OMIM #252900), tipo B (deficiência de alfa-N-acetilglicosamidase, OMIM #252920), tipo C (deficiência de acetil-CoA-alfa-glucosamina acetiltransferase, OMIM #252930) e tipo D (deficiência de N-acetilglicosamina 6-sulfatase, OMIM #252940)1. As mais comuns são as do tipo A e B, sendo as demais mais raras. Recentemente, identificou-se em modelo animal mais um subtipo, o E, contudo, até o momento não há pacientes humanos diagnosticados com essa forma de doença2,3.
A prevalência é de 1 para 70.000 nascidos vivos, sendo, portanto, o subtipo mais frequente de MPS quando comparados aos demais4. Os sintomas clínicos se iniciam entre 2 e 6 anos de idade1,4 e a expectativa de vida desses pacientes gira em torno de três a quatro décadas5. As manifestações clínicas da síndrome de Sanfilippo são principalmente problemas neurológicos1,4 (hiperatividade, distúrbios de sono, agressividade, atraso no desenvolvimento neuropsicomotor, dislalia, comportamento “autista-like”, etc.) e somáticos (infecções de ouvido e nariz de repetição, perda de audição, escoliose, lordose, hepatomegalia, etc.)4.
Este artigo relata o caso de uma paciente com o diagnóstico de MPS III-B, cujo diagnóstico precoce permitiu intervenções de reabilitação, que impactaram na qualidade de vida da paciente, e aconselhamento genético para seus genitores.
RELATO DE CASO
Paciente, sexo feminino, 5 anos, filha de pais jovens e não-consanguíneos, foi encaminhada para avaliação de distúrbio comportamental e atraso de linguagem.
Nasceu de parto cesárea eletivo, a termo (39 semanas), com 51cm de comprimento e peso de 2,7kg, sem relatos de intercorrências no período gestacional e neonatal. Os genitores negam outros agravos à saúde ou intercorrências clínicas da paciente; negam também outros familiares com sintomas semelhantes aos dela; a paciente possui dois irmãos mais velhos saudáveis.
A partir dos 2 anos de idade, os genitores perceberam que a criança apresentava dificuldade para formular frases completas, comportamento agitado, aparentemente sem demonstrar medo em situações de perigo (como atravessar uma rua com trânsito intenso de carros) e incoordenação motora. A paciente, contudo, demonstrava desenvolvimento motor adequado. Os genitores referiram que a pediatra observou hepatomegalia na paciente (confirmada por exame ultrassonográfico), regredindo posteriormente.
Paciente apresentava altura e peso adequados para a idade, sem sinais dismórficos evidentes, contudo, comparando a evolução das fácies da paciente em diferentes idades, pode-se considerar que houve aparecimento de traços faciais mais infiltrados (“coarsening facial features”) (Figura 1).
 Figura 1. Evolução do fenótipo facial da paciente. Fonte: Os autores (2018).
Figura 1. Evolução do fenótipo facial da paciente. Fonte: Os autores (2018).
Paciente frequentava escola normal e estava no ano competente à sua idade, porém, em vista das alterações de linguagem da paciente, o setor pedagógico da escola orientou a família a procurar tratamento fonoaudiológico para a paciente. Foi avaliada por um serviço de fonoaudiologia, que sugeriu a avaliação por um neuropediatra.
A avaliação com a neuropediatra sugeriu o diagnóstico de uma síndrome genética, interrogando uma forma de Sanfilippo (mucopolissacaridose do tipo III). No entanto, solicitou-se apenas o teste urinário para pesquisa de GAGs, cujo resultado foi dentro da normalidade (Tabela 1).
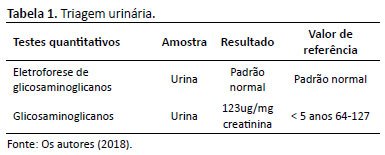
Posteriormente, a paciente foi encaminhada para serviço de genética médica, sendo levantada a hipótese de “forma leve/atenuada” de Sanfilippo. Iniciou-se o tratamento farmacológico para hiperatividade da paciente com clonazepam, e foram solicitados testes urinários e sanguíneos (testes enzimáticos) para a paciente. A triagem urinária para presença de GAGs e dosagem total de GAGs mostrou-se novamente dentro da normalidade (Tabela 1). No entanto, dada a suspeita clínica, procedeu-se ao estudo enzimático das quatro enzimas envolvidas na síndrome de Sanfilippo e observou-se a deficiência da enzima alfa-N-acetilglicosamidase (tipo B), confirmando o diagnóstico de Sanfilippo (Tabela 2).

No momento, paciente apresenta comportamentos desafiadores, como perda de medo e hiperatividade, com dificuldade de focar, mesmo que por alguns minutos, em uma única brincadeira ou atividade. Não havia sinais de autoagressividade, porém, a paciente, por vezes, exibia comportamento mais agressivo (como mordidas) voltadas para sua genitora, quando se sentia contrariada pela mesma. Além disso, apresentou até o momento velocidade de crescimento adequado para sua idade, embora com evidente estagnação de seu desenvolvimento neuropsicomotor sem, contudo, apresentar sinais de regressão neurológica. Genitores negam qualquer distúrbio do sono atual ou prévio.
DISCUSSÃO
Como a síndrome de Sanfilippo é uma doença que cursa com estagnação/regressão neurológica na infância, ela sempre deve estar presente como diagnóstico diferencial em crianças que desenvolvem transtornos de comportamentos5.
Tradicionalmente, a evolução da doença é dividida em 4 fases:
• Na fase pré-clínica, a criança possui um desenvolvimento neuropsicomotor normal. Em média, essa fase ocorre do nascimento até os dois anos de idade1,4,6.
• Na fase 1, a criança apresenta tanto distúrbios neurológicos (atraso no desenvolvimento neuropsicomotor ou atraso de fala) como problemas somáticos. Inicia-se por volta do primeiro até o quarto ano de vida4,7.
• Na fase 2, observa-se a presença de atraso no desenvolvimento associado a distúrbios do sono e comportamentos desafiadores (comportamento “autista-like”, perda de medo, agressividade e hiperatividade). Começa por volta do terceiro até o sétimo ano de vida4,6,7.
• Na fase 3, a criança geralmente fica menos agitada, refletindo, na realidade, o processo de demência associado à doença. Nela ocorre franca regressão, ainda que de forma lenta. Tem início por volta dos oito até os dez anos de idade4,6,8.
Em comparação com as outras mucopolissacaridoses que possuem um caráter mais multissistêmico, a MPS III pode se apresentar como uma doença puramente neurológica em vários pacientes. Isso decorre do tipo de GAG que se acumula nessa doença, no caso o heparan sulfato (Figura 2). O heparan sulfato se acumula no corpo do indivíduo de forma global em todos os órgãos, porém, diferente das outras MPSs, esse GAG é tóxico principalmente para as células do sistema nervoso central, levando ao processo de neurodegeneração4,9-11.
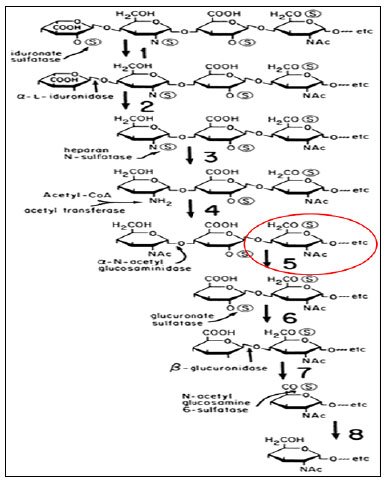 Figura 2. Rota metabólica GAG - Degradação gradual do heparan sulfato. As doenças de deficiência correspondem às reações numeradas: 1 = MPS II, Síndrome de Hunter; 2 = Síndromes de MPS I, Hurler, Hurler-Scheie e Scheie; 3 = MPS III-A, Síndrome de Sanfilippo tipo A; 4 = MPS III-C, Síndrome de Sanfilippo tipo C; 5 = MPS III-B, Síndrome de Sanfilippo tipo B; 6 = sem doença de deficiência ainda conhecida; 7 = MPS VII, Síndrome de Sly; 8 = MPS III-D, Síndrome de Sanfilippo tipo D. O desenho representa todas as estruturas conhecidas que ocorrem dentro do heparan sulfato, não implicando que elas ocorram estequiometricamente. Fonte: Bases of inherited disease (2014).
Figura 2. Rota metabólica GAG - Degradação gradual do heparan sulfato. As doenças de deficiência correspondem às reações numeradas: 1 = MPS II, Síndrome de Hunter; 2 = Síndromes de MPS I, Hurler, Hurler-Scheie e Scheie; 3 = MPS III-A, Síndrome de Sanfilippo tipo A; 4 = MPS III-C, Síndrome de Sanfilippo tipo C; 5 = MPS III-B, Síndrome de Sanfilippo tipo B; 6 = sem doença de deficiência ainda conhecida; 7 = MPS VII, Síndrome de Sly; 8 = MPS III-D, Síndrome de Sanfilippo tipo D. O desenho representa todas as estruturas conhecidas que ocorrem dentro do heparan sulfato, não implicando que elas ocorram estequiometricamente. Fonte: Bases of inherited disease (2014).
Na MPS III, o acúmulo do heparan sulfato nas células lisossomais gera um processo inflamatório cerebral extenso. Esse processo ocorre porque o heparan sulfato liberado pelos lisossomos são fagocitados pela micróglia. O mesmo acontece com os neurônios englobados e danificados pelo excesso desse glicosaminoglicano, iniciando um processo de alterações metabólicas nas células neurais (quando ocorre aumento de outros glicoesfingolipídeos como os gangliosídeos com potencial neurotóxico) que culmina com a ativação da apoptose (morte celular programada), levando à neurodegeneração12-14.

Apesar de constituírem o subtipo mais comum de MPS, ainda há bastante subdiagnóstico dessa doença, em virtude de pacientes com Sanfilippo não apresentarem o fenótipo clássico multissistêmico com “facies grosseiro” observado em outras MPSs, como a tipo I, tipo II e a tipo VI15.
Ao se fazer suspeita de MPS III, alguns exames são essenciais para definir a presença ou não da doença e, caso confirme, qual o subtipo. O primeiro exame complementar a ser feito é, usualmente, o exame de urina (cromatografia de glicosaminoglicanos para definir o tipo de GAG presente na urina e a dosagem total de GAGs na urina quantitativa) ao qual irá dizer de modo quantitativo a dose de heparan sulfato presente na urina do paciente1,15. Na síndrome de Sanfilippo, não é incomum o exame de urina ser negativo, porém, se a clínica do paciente for muito sugestiva, o clínico deve pedir o teste enzimático (feito a partir de amostra de sangue) para as quatro enzimas de MPS III para confirmar ou afastar esse diagnóstico1,15.
Além disso, é interessante ressaltar que crianças que adquiriram comportamento autista ou hiperativo, mas que não fecham critérios diagnósticos para autismo ou TDAH devem ser investigadas a MPS III como uma possível causa orgânica16-18.
A MPS III é uma doença que ainda não tem cura, mas existem terapias de reabilitação que visam desacelerar a progressão doença e dar uma melhor qualidade de vida ao portador da enfermidade19,20.
Assim, o benefício de um reconhecimento rápido da MPS III inclui aconselhamento genético para os genitores, início precoce de terapias de reabilitação e melhor qualidade de vida para o paciente e sua família21,22.
REFERÊNCIAS
1. National MPS Society. A guide to understanding MPS [Internet]. Durham: National MPS Society; 2018; [acesso em ANO Mês dia]. Disponível em: https://mpssociety.org/cms/wp-content/uploads/2017/04/booklet_MPS_III_v6.pdf
2. Bodamer OA, Giugliani R, Wood T. The laboratory diagnosis of mucopolysaccharidosis III (Sanfilippo syndrome): a changing landscape. Mol Genet Metab. 2014 Set/Out;113(1-2):34-41.
3. Fedele A. Sanfilippo syndrome: causes, consequences, and treatments. Dovepress. 2015;2015:269-81.
4. Jakobkiewicz-Banecka J, Gabig-Ciminska M, Kloska A, Malinowska E, Banecka-Majkutewicz Z, Banecki B, et al. Glycosaminoglycans and mucopolysaccharidosis type III. Front Biosci. 2016 Jun;21:1393-409.
5. Cohen MA, Stuart GM. Delivery of anesthesia for children with Mucopolysaccharidosis Type III (Sanfilippo syndrome): a review of 86 anesthetics. Pediatr Anesth. 2017Abr;27(4):363-9.
6. Wijburg FA, Wegrzyn G, Burton BK, Tylki-Szymanska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hypeeractivity disorder or autism spectrum disorder. Acta Pediatr. 2013 Mai;102(5):462-70.
7. Wolfenden C, Wittkowski A, Hare DJ. Symptoms of autism spectrum disorder (ASD) in individuals with mucopolysaccharide disease type III (Sanfilippo Syndrome): a systematic review. J Autism Dev Disord. 2017 Nov;41(11):3620-33.
8. Escolar ML, Jones SA, Shapiro EG, Horovitz DDG, Lampe C, Amartino H. Practical management of behavioral problems in mucopolysaccharidoses disorders. Mol Genet Metab. 2017;122(Supl 1):35-40.
9. Truxal KV, Fu H, McCarty DM, McNally KA, Kunkler KL, Zumberge NA, et al. A prospective one-year natural history study of mucopolisaccharidosis type IIIA and IIIB: Implications for clinical trial design. Mol Genet Metab. 2016 Nov;119(3):239-48.
10. Aoyagi-Scharber M, Crippen-Harmon D, Lawrence R, Vincelette J, Yogalingam G, Yip BK, et al. Clearance of heparan sulfate and attenuation of CNS pathology by intracerebroventricular BMN 250 in Sanfilippo type B mice. Am Soc Gene Cell Ther. 2017 Set;6:43-53.
11. King B, Hassiotis S, Rozaklis T, Beard H, Trim PJ, Snel MS, et al. Low-dose, continuous enzyme replacement therapy ameliorates brain pathology in the neurodegenerative lysosomal disorder mucopolysaccharidosis type III A. J Neurochem. 2016 Mai;107(3):409-22.
12. Martins C, Hulková H, Dridi L, Dormoy-Raclet V, Grigoryeva L, Choi Y, et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain. 2015 Fev;138(Pt 2):336-55.
13. Pshezhetsky AV. Lysosomal storage of heparan sulfate causes mitochondrial defects, altered autophagy, and neuronal death in the mouse model of mucopolysaccharidosis III type C. Autophagy. 12(6):1059-60.
14. Humano K, Hayashi M, Shioda K, Fukatsu R, Mizutani S. Mechanisms of neurodegeneration in mucopolysaccharidoses. Acta Neuropathol. 2008 Mai;115(5):547-59.
15. Valstar MJ, Ruijter GJG, Van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis. 2008 Abr;31(2):240-52.
16. Rio C, Machado RS, Pinheiro C, Eusébio F, Tasso T, Salgueiro E, et al. Perfil físico e psicológico de adolescentes e adultos com mucopolissacaridose sem atraso mental. Acta Pediatr. 1998;29(6):557-61.
17. Assumpção TM, Antúnez AEA. Avaliação do perfil psiquiátrico de pacientes com mucopolissacaridose [Internet]. São Paulo: Universidade de São Paulo (USP); 2013; [acesso em ANO Mês dia]. Disponível em: http://www.teses.usp.br/teses/disponiveis/47/47133/tde-24022014-094625/
18. Campos MG, Gonçalves AD. Relato de caso de clínico de mucopolissacaridose. Rev Int Odonto-Psicol. 2005;1(2):53-7.
19. Rossier VF, Guaré RO, Haddad AS, Ciamponi AL. Mucopolysaccharidosis III (Sanfilippo syndrome) – review and clinical case report. Rev Iberoam Odontopediatr Odontol Bebê. 2004;7(38):326-34.
20. Arita FN, Rosemberg S. Sindrome de sanfilippo (mucopolissacaridose III): estudo clinico das formas A e B. Rev Bras Neurol. 1988;24(5):135-8.
21. Lavery C, Hendriksz CJ, Jones S. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Out;12:168.
22. Demarco RC, Inada MS, Anselmo-Lima WT, Oliveira JAA, Feres MCC. Padrão auditivo em pacientes com Síndrome de Sanfilippo (Mucopolissacaridose tipo III-ß): relato de caso. São Paulo: SOB; 1997.
Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
Endereço para correspondência:
Charles Marques Lourenco
Centro Universitário Estácio de Ribeirão Preto
Rua Abrahão Issa Halach, 980 - Ribeirânia
Ribeirão Preto, SP, Brasil. CEP:14096-160
E-mail: charlesgenetica@gmail.com
Data de Submissão: 11/10/2019
Data de Aprovação: 11/05/2020
Recebido em: 11/10/2019
Aceito em: 11/05/2020
