Hipopituitarismo é a deficiência de um ou mais hormônios da glândula pituitária e pode surgir em consequência a uma variedade de desordens genéticas ou adquiridas1.
Complexas interações genéticas ditam o desenvolvimento normal da região hipotalâmica-hipofisária. Assim, mutações herdadas ou adquiridas podem ser responsáveis por deficiências hormonais congênitas ou hipopituitarismo de manifestação mais tardia, isoladas ou sindrômicas1, a exemplo da displasia septo-óptica2. Dentre as causas adquiridas, tumores afetando a região do hipotálamo-hipófise, danos locais secundários a trauma, irradiação e infecções são algumas das etiologias1.
As características clínicas do hipopituitarismo são variáveis tanto em severidade quanto dependente do número de hormônios afetados. A progressão de deficiências hormonais é característica bem estabelecida do hipopituitarismo, e por isso a presença de um eixo afetado (somatotrófico, gonadotrófico, tireotrófico ou corticotrófico) indica a avaliação dos demais no momento do diagnóstico e ao longo dos anos.
Este relato narrará um caso de hipopituitarismo em uma criança do sexo feminino, de 2 anos e 6 meses, no Distrito Federal, e tem por objetivo alertar o médico pediatra sobre os sintomas suspeitos, assim como repercussão da doença no desenvolvimento pôndero-estatural e neuropsicomotor de uma criança.
RELATO DE CASO
S.P.A., feminino, branca, 2 anos e 6 meses, apresentou em seu domicílio três episódios de crises convulsivas afebris. Levada ao serviço de emergência, glicemia 29mg/dL, feita correção desta com melhora da crise e internação para investigação.
Nascida a termo (39 semanas e 1 dia), parto cesáreo sem intercorrências, Peso 3.030g, Comprimento 48 cm, Apgar 8/9, adequada para idade gestacional (AIG). Exame físico sem alterações, incluso defeitos de linha média. Com 30h de vida, apresentou dificuldade para mamar, hipoatividade e hipotermia. Glicosímetro acusou low e criança necessitou de cuidados intensivos com infusão de glicose contínua por 72h. Não houve coleta de amostra crítica. Apresentou icterícia prolongada (até 1 mês e meio de vida) sem necessidade de fototerapia, sendo mãe e filha tipo sanguíneo A+. Teste do pezinho sem alterações. Desenvolvimento neuropsicomotor: sentou aos 9 meses, andou com 1 ano e 7 meses. Atualmente caminha, conversa e brinca muito. Nega internações prévias, cirurgias ou traumatismos.
Antecedentes familiares: pais não consanguíneos. Mãe: saudável, altura 159,7cm. Pai: saudável, altura 169,5cm. Irmãos saudáveis. Prima de 1º grau fez uso de hormônio de crescimento (GH) e possui alteração na fala.
À ectoscopia, apresenta fronte proeminente. Aparelhos cardiovascular, respiratório e abdominal sem alterações. Unhas e cabelos preservados. Peso: 9,05kg (z-escore: -2,97); Estatura: 80cm (z-escore: -3,07); IMC: 14,14 (z-escore: -1,27); estatura-alvo: 158,1cm.
Durante investigação inicial, obtiveram-se os seguintes resultados: TSH 2,66 (VR: 0,27-4,2); T4L: 0,891 (VR: 0,93-1,7); IGF1: 19 (VR: 18- 172); Cortisol: 12 (VR: 5,4-25). Interrogado hipopituitarismo e encaminhada ao serviço de endocrinologia pediátrica.
Realizada coleta de amostra crítica com 20h de jejum (glicemia de 45mg/dl), TSH 3,41 (VR 0,27-4,2), T4 L 0,797 (VR: 0,93-1,7), GH: 0,23(VR: >5), radiografia de mãos e punhos, a qual demonstrou idade óssea compatível com 3 meses (idade cronológica: 2 anos e 6 meses) e demais exames de triagem para baixa estatura e amostra crítica, normais. Ressonância Magnética (RM) de sela túrcica (Figura 1), que evidenciou sela rasa, haste hipofisária não identificada, hipossinal da adeno-hipófise e ausência de sinal da neuro-hipófise na sela, esses achados associados ao dados clínicos corroboram para o diagnóstico hipopituitarismo. Iniciado tratamento com levotiroxina 5mcg/kg/dia e somatropina 0,1UI/kg/dia.
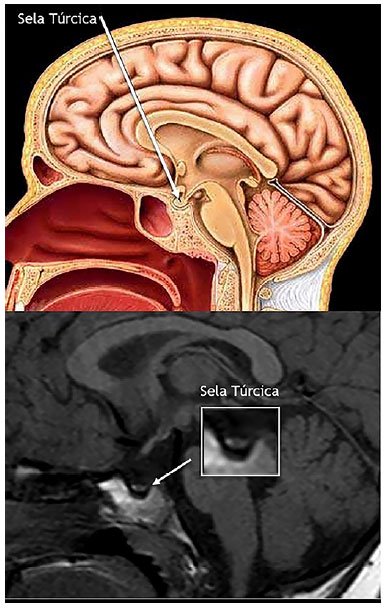 Figura 1. Desenho esquemático da sela túrcica. Logo abaixo, ressonância magnética ponderada em T1. Corte sagital demonstra sela rasa, hipossinal da adeno-hipófise e perda de sinal da neuro-hipófise. Ressonância magnética de sela túrcica que evidenciou sela rasa, haste hipofisária não identificada, hipossinal da adeno-hipófise e ausência de sinal da neuro-hipófise na sela. Os achados associados ao dados clínicos corroboram o diagnóstico de hipopituitarismo.
Figura 1. Desenho esquemático da sela túrcica. Logo abaixo, ressonância magnética ponderada em T1. Corte sagital demonstra sela rasa, hipossinal da adeno-hipófise e perda de sinal da neuro-hipófise. Ressonância magnética de sela túrcica que evidenciou sela rasa, haste hipofisária não identificada, hipossinal da adeno-hipófise e ausência de sinal da neuro-hipófise na sela. Os achados associados ao dados clínicos corroboram o diagnóstico de hipopituitarismo.A hipófise é uma glândula formada por vários tipos celulares, cujos produtos de secreção estimulam outras glândulas endócrinas periféricas a sintetizar e secretar hormônios envolvidos em funções diversas (crescimento, desenvolvimento neuropsicomotor, maturação sexual etc.)3. A região anterior da hipófise ou adeno-hipófise, de origem ectodérmica, produz o hormônio do crescimento (GH), as gonadotrofinas (LH e FSH), o hormônio estimulador da tireoide (TSH), o hormônio adrenocorticotrófico (ACTH) e prolactina. A região posterior ou neuro-hipófise, de origem neural, produz o hormônio antidiurético (ADH) e ocitocina4. Durante sua formação, com início entre 4ª - 5 ª semana de vida com a invaginação da bolsa de Rathke, há uma cascata temporalmente regulada de fatores de transcrição com a expressão dos hormônios da hipófise anterior. Conforme haja alteração nos fatores de transcrição, há deficiências hormonais mais frequentes conforme Figura 2 e Tabela 14.
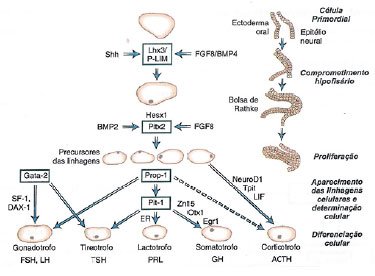 Figura 2. Modelo de desenvolvimento da glândula da hipófise anterior humana e da determinação da linhagem celular através da cascata de fatores de transcrição. O Lhx3 determina a diferenciação das células GH, Prolactina e TSH. O PTx se comporta como regulador hipofisário universal. GATA-2 regula a expressão da subunidade alfa presente nas gonadotrofinas e TSH. O PROP-1 é o principal determinante das linhagens celulares hipofisárias derivadas das células-tronco totipotenciais, que determina a expressão do PIT-1 e células gonadotróficas. A expressão do PIT-1 determina o desenvolvimento de GH, prolactina, TSH e do receptor do GH.
Figura 2. Modelo de desenvolvimento da glândula da hipófise anterior humana e da determinação da linhagem celular através da cascata de fatores de transcrição. O Lhx3 determina a diferenciação das células GH, Prolactina e TSH. O PTx se comporta como regulador hipofisário universal. GATA-2 regula a expressão da subunidade alfa presente nas gonadotrofinas e TSH. O PROP-1 é o principal determinante das linhagens celulares hipofisárias derivadas das células-tronco totipotenciais, que determina a expressão do PIT-1 e células gonadotróficas. A expressão do PIT-1 determina o desenvolvimento de GH, prolactina, TSH e do receptor do GH.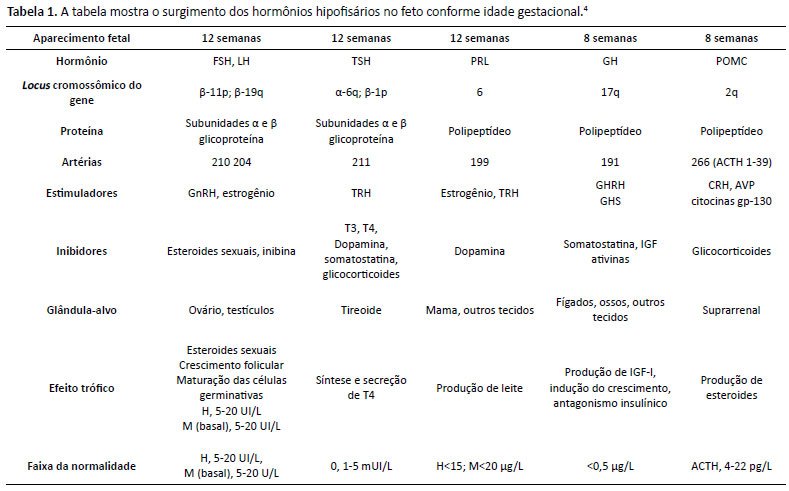
A apresentação clínica do hipopituitarismo pode incluir baixa estatura, hipotireoidismo, distúrbios de maturação sexual, hipoglicemia e sinais e sintomas de hipocortisolismo, isolados ou simultâneos5. Ausência congênita da glândula hipófise (aplasia), hipoplasia parcial ou rudimentos teciduais ectópicos raramente são encontrados5. Destaca-se que este último foi o observado na paciente em questão.
O comprometimento da síntese de um ou mais hormônios hipofisários pode resultar de fatores genéticos hereditários (mutação de genes como PROP-1, POU1F1, HESX1, LHX3 e T-Pit; síndrome de Laurence-Moon-Biedl, síndrome de Prader Willi e síndrome de Kallmann) e de lesões adquiridas (tumor, trauma, cirurgia ou irradiação)4,5. A literatura demonstrou recentemente uma nova mutação no gene PROP-15 associada à deficiência de GH e TSH e ao aumento da glândula pituitária, representando a mutação mais frequente no pan-hipopituitarismo, sendo 11% dos casos globais; 6,7% esporádicos e 48,5% familiares. Nesse caso, uma criança italiana, branca, de 3 anos, apresentou ao nascimento icterícia e níveis glicêmicos normais. Na idade de 3 anos, observou-se um declínio linear de seu crescimento, associado à obesidade truncal, "rosto de boneca" e fronte proeminente, porém sem atraso do desenvolvimento neuropsicomotor. Aos 3 anos, sua altura era de 78,7cm (- 4,23 desvios-padrão) e seu peso era de 10,8kg (-2,6 desvios-padrão). Idade óssea: 1 ano e 2 meses. Após confirmação de deficiência dos eixos tireotrófico e somatotrófico com exames laboratoriais, iniciou-se a reposição hormonal5.
O hipopituitarismo congênito é, também, uma causa incomum de colestase neonatal e pouco se sabe dos efeitos hormonais da pituitária anterior nas funções hepáticas. Na literatura, um artigo6 cita 8 casos de recém-nascidos que desenvolveram icterícia colestática e posteriormente deficiência hormonal adeno-hipofisária. Sinais como hipoglicemia, anormalidades oculares e micropênis foram achados frequentes6. Um ou mais defeitos no ACTH, GH e TSH têm sido relatados como responsáveis pela colestase; todavia, outras publicações relatam somente o cortisol como causa de colestase, não havendo consenso7.
Os pacientes com insuficiência hipofisária, a despeito da sua história, apresentam alto índice de mortalidade, principalmente por doença respiratória e vascular. A idade do diagnóstico, o sexo feminino e uma história de craniofaringioma são os fatores mais evidentes relacionados ao aumento da mortalidade4. Outro caso na literatura8 que demonstra a possível gravidade do hipopituitarismo relata história de recém-nascido, masculino, nascido de 41 semanas e peso de 3.320g, com desconforto respiratório após 1 hora do nascimento, necessidade de ventilação mecânica, seguindo-se choque cardiogênico refratário a catecolaminas. Hipopituitarismo severo foi considerado devido ao distúrbio respiratório, hipoglicemia e presença de micropênis. Após administração de hidrocortisona, houve melhora relevante da hemodinâmica. Sua ressonância magnética demonstrou aplasia da adeno-hipófise e glândula posterior ectópica8.
Ao se analisar cada eixo hipofisário, é constatado que a deficiência do hormônio de crescimento (DGH) é a deficiência adeno-hipofisária mais frequente nos portadores de doenças hipotálamo-hipofisárias4. A investigação para DGH está indicada quando há: 1) baixa estatura grave; 2) baixa estatura associada à redução na velocidade de crescimento; 3) estatura acima de -2 desvios-padrão para a idade e sexo, associada a uma baixa velocidade de crescimento; 4) presença de condição predisponente, como lesão intracraniana e irradiação do sistema nervoso central (SNC); 5) deficiência de outros hormônios hipofisários; e 6) sinais e sintomas de DGH/hipopituitarismo no período neonatal (hipoglicemia, icterícia prolongada, micropênis, defeitos de linha média)9. História de parto traumático, consanguinidade, outro membro da família afetado e presença de outras deficiências hormonais, também devem levantar a hipótese de DGH3.
Em relação à hipoglicemia, merecem investigação as seguintes situações: 1) hipoglicemia em RN saudável; 2) hipoglicemia recorrente ou persistente (>48h de vida); 3) necessidade de velocidade de infusão de glicose (VIG) >14 e 4) associação com outras anormalidades10. A investigação laboratorial deve ser iniciada durante o atendimento da crise hipoglicêmica. A obtenção de uma amostra crítica de sangue e urina durante a hipoglicemia é de extrema importância para seu diagnóstico e tratamento11.
O hipotireoidismo congênito classifica-se em primário, quando a alteração ocorre na glândula tireoide; secundário, quando ocorre na hipófise, e terciário, quando ocorre no hipotálamo; estes dois últimos caracterizam o "hipotireoidismo central"12. As principais consequências da ausência dos hormônios tireoidianos são: retardo mental grave, falência do crescimento, ataxia, incoordenação motora, estrabismo, movimentos coreiformes e perda auditiva neurossensorial12. No Brasil, a dosagem de TSH por imunofluorimetria, em amostra de sangue coletada em papel-filtro é a rotina recomendada para a triagem neonatal do hipotireoidismo congênito, sendo que valores de TSH acima de 10U/ml sinalizam para triagem positiva13. A paciente do presente relato apresentou, entretanto, "teste do pezinho" sem alterações, o que descartou hipotireoidismo congênito primário, porém não central. Estudo prospectivo da Argentina14 demonstrou que a adição de rotina de T4 na estratégia de rastreio neonatal pode diagnosticar com sucesso o hipotireoidismo congênito central - que foi responsável, nesse estudo, por 11% dos casos, em amostra de 22.573 recém-nascidos. A importância desse diagnóstico está na relação, na maioria dos casos, do hipotireoidismo central com déficits hormonais da hipófise potencialmente fatais (insuficiência adrenal e deficiência de hormônio do crescimento), como relatado14.
Em crianças, a deficiência dos hormônios gonadotróficos pode apresentar ausência de desenvolvimento puberal, atraso na maturação óssea e, nos meninos, micropênis e criptorquidia3.
Na hipófise, as células produtoras de ACTH estão entre as mais resistentes, sendo uma das últimas a terem sua função comprometida quando da presença de tumores, traumas (cirúrgicos ou não) e radioterapia nessa região. Os principais sinais e sintomas que sugerem a deficiência de ACTH são: anorexia, náuseas, vômitos, perda de peso, astenia, palidez cutânea, diminuição de pelos axilares e pubianos3.
Diante do exposto, a paciente do presente caso - a qual vem sendo acompanhada ambulatorialmente e com boa evolução - apresenta uma doença que representa um desafio diagnóstico, necessitando de tratamento imediato e em longo prazo para evitar complicações. A realização de investigação para mutações genéticas não foi possível devido à falta de insumos.
REFERÊNCIAS
1. Brook CGD, Clayton PE, Brown RS. Congenital Disorders of the Hypothalamo-Pituitary-Somatotrope Axis. In: Mehta A, Gevers&Mehul ETTD, orgs. Brook's Clinical Pediatric Endocrinology. 6th ed. Oxford: John Wiley & Sons, Ltd; 2009. p. 60-7.
2. Fernandes SY, Gray MCF, Dyonisio ACS. Síndrome de Morsier: relato de dois casos na cidade de Petrópolis. Resid Pediatr. 2020;10(1):44-47.
3. Sociedade Brasileira de Endocrinologia e Metabologia. Projeto Diretrizes - Hipopituitarismo: Diagnóstico. 2006; [acesso em 2019 Set 29];13. Disponível em: https://diretrizes.amb.org.br/_BibliotecaAntiga/hipopituitarismo-diagnostico.pdf.
4. Melmed S, Kleinberg D. Hipófise Anterior, Fatores de Transcrição Hipofisários. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, orgs. Williams Tratado de Endocrinologia. 11ª ed. Rio de Janeiro: Elsevier; 2010. p. 136-7.
5. Penta L, Bizzarri C, Panichi M, Novelli A, Lepri FR, Cappa M, et al. Identification of a novel PROP1 mutation in a patient with combined pituitary hormone deficiency and enlarged pituitary. Int J Mol Sci. 2019;20(8):1-7.
5. Karnsakul W, Sawathiparnich P, Nimkarn S, Likitmaskul S, Santiprabhob J, Aanpreung P. Anterior pituitary hormone effects on hepatic functions in infants with congenital hypopituitarism. Ann Hepatol. 2007;6(2):97-103.
6. Wada K, Kobayashi H, Moriyama A, Haneda Y, Mushimoto Y, Hasegawa Y, et al. A case of an infant with congenital combined pituitary hormone deficiency and normalized liver histology of infantile cholestasis after hormone replacement therapy. Clin Pediatr Endocrinol. 2017;26(4):251-7.
7. Ueda Y, Aoyagi H, Tajima T. A newborn with combined pituitary hormone deficiency developing shock and sludge. J Pediatr Endocrinol Metab. 2017;30(12):1333-6.
8. Ministério da Saúde (BR). Protocolo Clínico e Diretrizes Terapêuticas para Deficiência de Hormônio do Crescimento - Hipopituitarismo. Portal Arquivos [Internet]. 2018; [acesso em 2019 Set 29]. Disponível em: http://portalarquivos2.saude.gov.br/images/pdf/2018/dezembro/14/PCDT-Deficiencia-do-Hormonio-de-Crescimento-Hipopituitarismo.pdf.
9. Universidade Federal do Triângulo Mineiro. Hospital de Clínicas da UFTM. Protocolo Clínico (PC): "Hipoglicemia Neonatal - Condutas Médicas". 2019; [Acesso em 29 setembro 2019]. Uberaba: EBSERH, 2019. Disponível em: http://www2.ebserh.gov.br/documents/147715/0/Hipoglicemia+neonatal++vers%2Búo+final.pdf/10e7a2ba-8c7c-4b2a-870d-241da35b04db
10. Sociedade Brasileira de Pediatria. Diretrizes SBP - Hipoglicemia no Período Neonatal. SBP [Internet]. 2014; [acesso em 2021 Fev 20];(1). Disponível em: https://www.sbp.com.br/fileadmin/user_upload/2015/02/diretrizessbp-hipoglicemia2014.pdf.
11. Cristina LP de P, Garcia LS, Collett-Solberg PF, Liberatore Del Roio R, Pinto RM, Arrais RF, et al. Hipotireoidismo congênito: triagem neonatal. Soc Bras Pediatr. 2018;1-12.
13. Ministério da Saúde (BR). Protocolo clínico e diretrizes terapêuticas para hipotireoidismo congênito. Portaria SAS/MS no 1161, de 18 de novembro de 2015 [Internet]. 2015; [acesso em 2019 Set 29];1-8. Disponível em: http://portalarquivos.saude.gov.br/images/pdf/2015/novembro/26/Hipotiroidismo-congenito---PCDT-Formatado--.pdf.
14. Braslavsky D, Mendez MV, Prieto L, et al. Pilot neonatal screening program for central congenital hypothyroidism: evidence of significant detection. Horm Res Paediatr. 2017;88(3-4):274-80.
1. Hospital Regional de Ceilândia, Pediatria - Brasília - Distrito Federal - Brasil
2. Hospital Regional da Asa Norte, Pediatria - Brasília - Distrito Federal - Brasil
3. Hospital Regional de Ceilândia, Clínica Médica - Brasília - Distrito Federal - Brasil
4. Ambulatório Médico de Especialidades, Endocrinologia Pediátrica - Votuporanga - São Paulo - Brasil
Endereço para correspondência:
Anna Paula Cesar Costa
Hospital Regional de Ceilândia
QNM 27 Área Especial 1, QNM 28, Ceilândia
Brasília, DF, Brasil. CEP: 72215-274
E-mail: annapaulacesar@gmail.com
Data de Submissão: 02/03/2021
Data de Aprovação: 06/07/2021
Recebido em: 02/03/2021
Aceito em: 06/07/2021
