INTRODUÇÃO
A hipoglicemia persistente é uma condição desafiadora, sendo a hiperinsulinemia a principal causa de hipoglicemia persistente e recorrente na infância. Descrevemos um caso de hipoglicemia grave em uma lactente com estigmas sindrômicos diagnosticada com Síndrome de Shashi-Pena após teste genético. Como há variabilidade na apresentação e gravidade dos sintomas das pessoas com essa síndrome, o presente relato de caso tem como finalidade apresentar e auxiliar na compreensão da síndrome de Shashi-Pena e no tratamento da hipoglicemia hiperinsulinêmica.
RELATO DE CASO
Lactente de 2 anos, sexo feminino, filha de pais caucasianos e não consanguíneos. Mãe primípara de 19 anos, apresentou hipotireoidismo gestacional tratada com levotiroxina. Não houve consumo de álcool, substâncias ilícitas e tabaco durante a gestação. Como na última ecografia fetal do pré-natal foi evidenciada restrição do crescimento intrauterino grau 2, optou-se por interromper a gestação por meio de parto cesárea. A paciente nasceu com peso 1985g (Z escore -2,77), comprimento 42,5cm (Z escore -3,16) e perímetro cefálico 32cm (Z escore -1,13), em bom estado geral, sem necessidade de reanimação neonatal. Ao exame físico, verificou-se a presença de nevus flammeus na glabela, sobrancelhas arqueadas e longas, sinofris, lábio superior fino, palato ogival, frênulo sublingual, micrognatia, sulcos plantares profundos, hipertricose lanuginosa generalizada e hipotonia axial. Devido ao baixo peso ao nascer e um episódio assintomático de hipoglicemia, foi encaminhada à Unidade de Terapia Intensiva neonatal. A figura 1 demonstra a paciente com síndrome de Shashi-Pena logo após o nascimento.
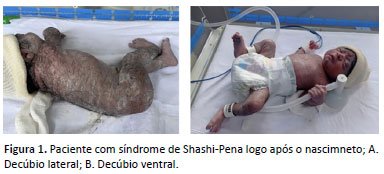
No período neonatal, apresentou importante dificuldade alimentar via oral devido à incoordenação de sucção-deglutição-respiração, com necessidade de sondagem. Evoluiu com episódios recorrentes de hipoglicemia, com necessidade de uma taxa de infusão máxima de 12mg/kg/min para manter a glicemia dentro da normalidade. Apresentou, ainda, diarreia persistente, sendo necessário introduzir fórmula infantil extensamente hidrolisada, com boa resposta após.
Devido recém-nascido com restrição de crescimento uterino, pequeno para idade gestacional e com episódios recorrentes de hipoglicemias, foram realizados exames de imagem com os seguintes resultados: potencial Evocado Auditivo de Tronco Encefálico (alteração auditiva bilateral); ecocardiograma com doppler (persistência do canal arterial e comunicação interatrial pequena); ultrassonografia do aparelho urinário (rins em ferradura); ressonância magnética do crânio (hipoplasia do vérmis cerebelar e dilatação do quarto ventrículo, achados sugestivos de malformação de Dandy-Walker); eletroencefalograma (atividade epileptiforme frequente localizada nas regiões fronto-centro-temporais).
Como a hipoglicemia persistiu além do período neonatal, foi optado por iniciar prednisolona 2,5 mg/m2 diária, inicialmente isolada e posteriormente associada ao GH, porém sem melhora. Aos 8 meses de idade, foi possível coletar amostra crítica durante um episódio de glicemia <50mg/dL e realizado teste de estimulação com glucagon (importante para o diagnóstico da hipoglicemia hiperinsulinêmica). Aos 12 meses de idade, foi possível iniciar o medicamento diazóxido (na dose de 5mg/Kg/dia), evoluindo com adequado controle glicêmico.
Diante desses achados, foi realizada a análise molecular por sequenciamento de exoma e identificada a variante patogênica chr2:25.749.716 G>A, em heterozigose no gene ASXL2 confirmando condição geneticamente determinada conhecida como Síndrome de Shashi-Pena (OMIM # 617190).
A paciente apresentou infecções respiratórias de repetição (aos 4, 7, 10, 12, 13 e 15 meses), sendo necessário internamento em enfermaria pediátrica em 4 ocasiões. Devido ao padrão radiológico mantido no lobo superior direito entre os episódios, foi iniciada investigação de imunodeficiência: hemograma completo normal, dosagem de imunoglobulinas dentro dos percentis para a idade e imunofenotipagem por citometria de fluxo com diminuição da contagem de linfócitos B CD19+CD20+ e de linfócitos NK CD16+CD56+. Por ora, mantido sem medicação.
Realizado ecocardiograma com doppler controle com 1 ano e 3 meses de idade: persistência do canal arterial, átrio esquerdo aumentado e ventrículo esquerdo globoso. Encaminhada ao serviço de referência, onde foi indicado cateterismo cardíaco, sem necessidade de tratamento farmacológico.
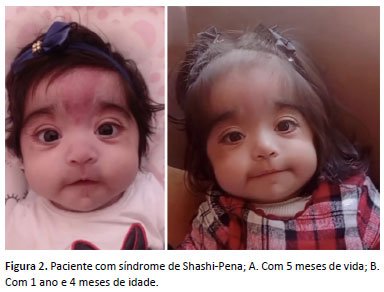
Durante os dois primeiros anos de vida, observou-se a não recuperação espontânea do crescimento (PIG sem catch up). Houve também atraso global do desenvolvimento: não senta sem apoio, não fala palavras e não se alimenta sozinha. Atualmente, a paciente mantém acompanhamento multidisciplinar e realiza terapias complementares semanalmente. A figura 2 demonstra a paciente com síndrome de Shashi-Pena: A) Com 5 meses de vida; B) Com 1 ano e 4 meses de idade.
DISCUSSÃO
A síndrome de Shashi-Pena é decorrente de variantes patogênicas em heterozigose no gene ASXL2 e é uma condição geneticamente determinada que pode ser de herança autossômica dominante ou secundária a um evento mutacional "de novo", ou seja, uma variante que surge em um indivíduo pela primeira vez e não foi herdada de um dos pais1,2.
Estima-se uma prevalência de 45 casos da síndrome em todo o mundo. A síndrome de Shashi-Pena foi descrita na literatura pela primeira vez em 2016 e é caracterizada por dismorfismos faciais (macrocefalia, nevus flammeus na região da glabela, hipertelorismo e sobrancelhas arqueadas), anormalidades cardíacas congênitas, hipoglicemia, dificuldades alimentares, alterações na densidade mineral óssea, convulsões, hipotonia e atraso no desenvolvimento neuropsicomotor1-4.
A paciente deste caso apresenta dismorfismos e alterações fenotípicas compatíveis com a síndrome de Shashi-Pena, contudo não apresentou macrocefalia e foi um recém-nascido pequeno para a idade gestacional (por peso e comprimento), o que vai contra o relatado na literatura atual1-4.
Um achado importante neste caso é o diagnóstico de hipoglicemia hiperinsulinêmica congênita. O hiperinsulinismo pode ser transitório (<6 meses) ou persistente (>6 meses) e é a principal causa de hipoglicemia persistente e recorrente na infância. Quando iniciada no período neonatal e mantida posteriormente, classifica-se como forma congênita5-7.
Recém-nascidos pequenos para a idade gestacional (PIG), com restrição do crescimento intrauterino (RCIU) e/ou sinais sugestivos de síndrome genética têm maior risco de apresentar hipoglicemia neonatal devido à alteração em alguma via metabólica (glicogenólise, gliconeogênese, oxidação de ácidos graxos mitocondriais ou cetogênese)6,7. Os sinais e sintomas de hipoglicemia são variados e incluem agitação, irritabilidade, palidez, cianose, hipotermia, letargia, apneia e crise convulsiva7.
Na investigação da hipoglicemia neonatal (dosagem plasmática inferior à 50mg/dL), deve ser coletada no momento da hipoglicemia uma amostra dos seguintes exames de sangue: glicose plasmática, gasometria (arterial ou venosa), lactato, eletrólitos (sódio, potássio e cloro), insulina, amônia, cortisol, GH, peptídeo-C, beta-hidroxibutirato, ácidos graxos livres, perfil de acilcarnitinas e de aminoácidos. É importante coletar concomitantemente um parcial de urina para avaliar a presença de cetonúria e/ou de substâncias redutoras6,7.
Se a hipoglicemia cursar com acidose metabólica, deve-se investigar glicogenoses, distúrbios da gliconeogênese e hipopituitarismo. Se a hipoglicemia ocorrer sem acidose metabólica, investiga-se hiperinsulinismo e distúrbios da oxidação de ácidos graxos6,7.
É essencial determinar a causa da hipoglicemia, pois o tratamento difere conforme a doença de base. No caso da hipoglicemia hiperinsulinêmica, o medicamento de escolha é o diazóxido8. Um análogo da somatostatina (octreotida) deve ser considerado para pacientes com hipoglicemia refratária, como relatado em um caso chinês9.
CONCLUSÃO
Existe variabilidade de sinais e sintomas entre pacientes diagnosticados com uma mesma síndrome genética. O presente relato de caso tem como principal finalidade contribuir com o espectro de fenótipos e variações genéticas da síndrome de Shashi-Pena. Desde 2016, apenas 13 casos foram relatados na literatura. Esta é a primeira vez que o diazóxido foi administrado em um paciente com hipoglicemia persistente secundária à síndrome de Shashi-Pena, ajudando a manter um controle glicêmico adequado.
REFERÊNCIAS
1. Shashi V, Pena LDM, Kim K, Burton B, Hempel M, Schoch K, et al. De Novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am J Hum Genet [Internet]. 2017 Jan 5; [cited 2024 Feb 17]; 100(1):179. DOI: https://doi.org/10.1016/j.ajhg.2016.12.004. Available from: https://pubmed.ncbi.nlm.nih.gov/27693232/.
2. Cuddapah VA, Dubbs HA, Adang L, Kugler SL, McCormick EM, Zolkipli-Cunningham Z, et al. Understanding the phenotypic spectrum of ASXL -related disease: Ten cases and a review of the literature. Am J Med Genet A [Internet]. 10 mar 2021; [cited 2024 Feb 17]. DOI: https://doi.org/10.1002/ajmg.a.62156. Available from: https://pubmed.ncbi.nlm.nih.gov/33751773/.
3. Wang Y, Tan J, Wang Y, Liu A, Qiao F, Huang M, et al. Diagnosis of Shashi-Pena Syndrome caused by chromosomal rearrangement using nanopore sequencing. Neurol Genet [Internet]. 23 nov 2021; [cited 2024 Feb 17]; 7(6):e635. DOI: https://doi.org/10.1212/nxg.0000000000000635. Available from: https://pubmed.ncbi.nlm.nih.gov/34841066/.
4. Liberatore Junior RD, Negri AA, Martinelli Junior CE, Kochi C, Silva IN, Collett-Solberg PF. Hipoglicemia hiperinsulinêmica da infância: Análise de dados clínicos de uma amostra brasileira. Arq Bras Endocrinol Amp Metabol [Internet]. 2012 Dez; [citado 2024 Fev 17]; 56(9):666-71. DOI: https://doi.org/10.1590/s0004-27302012000900011. Disponívl em: https://www.scielo.br/j/abem/a/H5wLBKy8YfMXH9vMXP3frJp/.
5. Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis [Internet]. 2011; [cited 2024 Feb 17]; 6(1):63. DOI: https://doi.org/10.1186/1750-1172-6-63. Available from: https://pubmed.ncbi.nlm.nih.gov/21967988/.
6. Marinho PC, Sá AB, Gouveia BM, Serpa JB, Moraes JRS, Sodré RS, et al. Hipoglicemia neonatal: revisão de literatura. Braz J Health Rev [Internet]. 2020; [citado 2024 Fev 17]; 3(6):16462-74. DOI: https://doi.org/10.34119/bjhrv3n6-068. Disponível em: https://ojs.brazilianjournals.com.br/ojs/index.php/BJHR/article/view/20050.
7. Pediatric Endocrinology. Williams textbook of endocrinology. Moscow: GEOTAR-Media; Publishing Group eBooks; 2020.
8. Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol [Internet]. 2010 May; [cited 2024 Feb 17]; 162(5):987-92. DOI: https://doi.org/10.1530/eje-09-0861.
9. Yuan M, Shan Y, Xu F, Yang L, Sun C, Cheng R, et al. A newborn with a pathogenic variant in ASXL2 expanding the phenotype of SHAPNS: a case report and literature review. Transl Pediatr [Internet]. 2023 Jan; [cited 2024 Feb 17]: 0. DOI: https://doi.org/10.21037/tp-22-220. Available from: https://pubmed.ncbi.nlm.nih.gov/20164212/.
1. Hospital Universitário Evangélico Mackenzie, Departmento de Paediatria - Curitiba - Paraná - Brasil
2. Faculdade Evangélica Mackenzie do Paraná, Medicina - Curitiba - Paraná - Brasil
Endereço para correspondência:
Carolina Oliveira De-Paulo
Hospital Universitário Evangélico Mackenzie, Department of Paediatric - Curitiba - Paraná - Brasil
Alameda Princesa Izabel, 1585, Bigorrilho
Curitiba, PR, Brasil. CEP: 80730-080
E-mail: loracoliveira@hotmail.com
Data de Submissão: 03/07/2023
Data de Aprovação: 13/03/2024
Recebido em: 03/07/2023
Aceito em: 13/03/2024
