INTRODUÇÃO
A lipodistrofia de Berardinelli, ou Síndrome de Berardinelli-Seip (SBS), é uma patologia rara, tendo como alterações fenotípicas a presença de redução ou ausência de tecido adiposo e hipertrofia do músculo esquelético1. Essa enfermidade costuma ser identificada clinicamente pela presença de características físicas marcantes como aspecto acromegaloide, face triangular com bochechas profundas pela perda da gordura de Bichat, musculatura hipertrofiada, hepatomegalia, protrusão da cicatriz umbilical e proeminência dos vasos sanguíneos (flebomegalia)2. Essa condição foi descrita pela primeira vez pelo médico brasileiro Berardinelli, em 1954, sendo uma doença genética rara pertencente ao grupo das lipodistrofias, caracterizadas pela ausência ou redução anormal de tecido adiposo no corpo3. Objetivos deste relato: (1) alertar os médicos sobre essa patologia e suas consequências no cotidiano do paciente; (2) conscientizar sobre a importância do diagnóstico precoce, mesmo sem os testes moleculares, e seu impacto na redução de comorbidades relacionadas a essa síndrome.
RELATO DE CASO
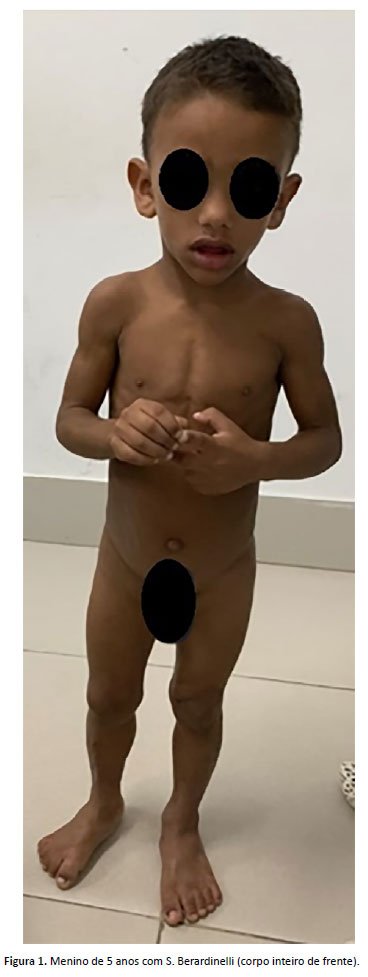
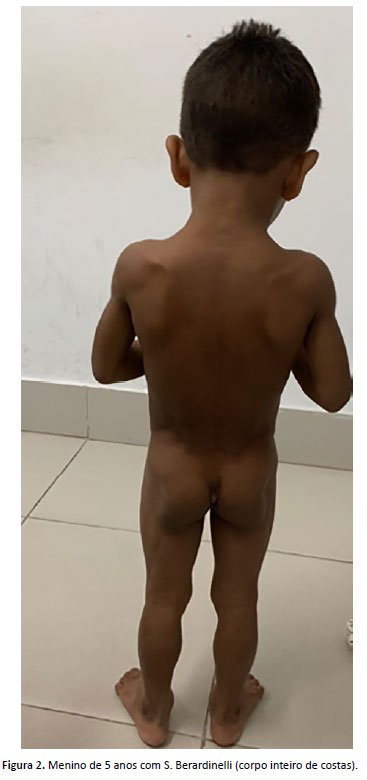
FDSS, menino com 5 anos, atendido no ambulatório de genética de um hospital público de referência para tratamento de doenças raras do Ceará apresentava dismorfias faciais que foram o motivo da consulta. Os pais eram consanguíneos. A mãe tinha 21 anos e o pai tinha 42 anos, sem outros casos na família. Nasceu de parto normal, sentou com 6 meses, andou com 1 ano, falou com 2 anos e controlou esfíncteres com 1 ano e 3 meses. Não houve problemas escolares. Havia relato de agitação e história de hiperfagia. Medidas exame físico: peso = 14,4 kg (Z score -3 e -2), estatura = 100 cm (Z score -2); perímetro cefálico = 51cm (Z score -2 e 0), havia fácies com escassez de tecido adiposo, membros com musculatura hipertrofiada, lipodistrofia, plicoma anal, pés planos (Figura 1 e 2). Foram realizados: tomografia de crânio, ultrassonografia abdominal e radiografia de esqueleto total, que foram normais. Dosagem de triglicerídeos foi 218 mg/dL (VR<75 mg/dL (entre 0 e 9 anos). Presença de três critérios maiores de diagnóstico, como lipodistrofia que afeta tronco, membros e face; aparência acromegaloide; elevada concentração sérica de triglicerídeos (>80 g/L), foi feito o diagnóstico clínico de SBS e instituído o manejo do caso. Foi possível realização do painel de genes para hipertrigliceridemia, que demonstrou mutação no gene AGPAT2 (1-acylglycerol-3-phosphate O-acyltransferase 2). Atualmente o paciente encontra-se em acompanhamento pelo geneticista, endocrinologista e nutricionista, tendo uma abordagem multidisciplinar em seu seguimento ambulatorial. Foi realizado aconselhamento genético.
DISCUSSÃO
A SBS é uma doença rara cuja incidência pode variar dependendo da população, sendo 1:1.000.000 nos EUA e 1:500.000 em Portugal4. Não temos dados epidemiológicos no Brasil, mas, como brasileiros têm ascendência étnica portuguesa, deve-se ter uma incidência aproximada a de Portugal. Além disso, há rumores da existência de um cluster no Rio Grande do Norte, estado vizinho do Ceará, onde reside o paciente. O caso que apresentamos se trata de um paciente com suspeita clínica da SBS, confirmada por exames laboratoriais. Consideramos que o diagnóstico clínico dessa doença deve ser enfatizado para que medidas terapêuticas sejam instituídas precocemente.
O diagnóstico pode ser dado a partir da identificação de
• três critérios maiores ou
• dois critérios maiores e dois ou mais critérios menores, ou
• identificação das variantes patogênicas bialélicas no gene AGPAT25.
Dentre os critérios maiores, destacam-se (1) presença de lipodistrofia que afeta tronco, membros e face; (2) aparência acromegaloide; (3) hepatomegalia; (4) elevada concentração sérica de triglicerídeos (>80 g/L) e (5) resistência à insulina5. Para os menores, (1) cardiomiopatia hipertrófica; (2) retardo de desenvolvimento neurológico ou intelectual leve a moderado; (3) hirsutismo/hipertricose; (4) puberdade precoce feminina; (5) cistos ósseos e (6) flebomegalia5. No caso que apresentamos, havia três critérios maiores (1,2,4), que nos fizeram realizar o diagnóstico clínico dessa doença, sendo importante para evitarmos complicações e iniciar o tratamento adequado, evitando dislipidemia, efeitos da resistência à insulina e cardiopatia.
O acúmulo de gordura visceral ao redor dos órgãos internos, especialmente no fígado, levando à hepatomegalia, pode contribuir para resistência à insulina e desenvolvimento de diabetes mellitus tipo 2, que deve ser evitado ao máximo, por isso a presença do endocrinologista e da nutricionista na equipe multidisciplinar que vai cuidar desse paciente é importante6.
A SBS é causada por mutações em genes específicos que desempenham papel crucial no desenvolvimento e regulação do tecido adiposo, destacando-se as alterações no gene AGPAT2. Este está envolvido na síntese de lipídios, desempenhando um papel fundamental na formação do tecido adiposo, logo suas alterações levam a anormalidades na produção e distribuição de gordura no corpo, resultando na característica falta de tecido adiposo subcutâneo7. Nos casos típicos, o diagnóstico pode ser feito pelos critérios supracitados, porém o teste molecular é importante para a definição etiológica nos casos de fenótipo incompleto e para determinação de diagnóstico pré-natal de outros membros da família, podendo utilizá-lo no aconselhamento genético. No entanto, não se recomenda esperar pelo resultado para iniciar as intervenções terapêuticas, pois são de alto custo e podem ser de difícil acesso para pacientes atendidos na rede pública de saúde.
No caso que descrevemos, os pais eram consanguíneos, o que predispõe a ocorrência de doenças autossômicas recessivas, como a SBS, porém, como no nordeste brasileiro a consanguinidade é muito frequente, não podemos definir que todo casal consanguíneo teria filhos com doenças genéticas de herança autossômica recessiva8. Para o caso atual, orientou-se a família sobre a doença e recebeu risco de recorrência de 25%, podendo a mutação já identificada ser utilizada para diagnóstico de certeza para outros casos, caso desejem.
O manejo clínico da SBS consiste em estratégias para lidar com suas complicações metabólicas, sendo o gerenciamento da dieta o aspecto mais importante no controle de efeitos biológicos, restringindo a ingestão total de energia, gordura saturada e carboidratos simples, dando preferência ao consumo de carboidratos complexos, fibras solúveis, triglicerídeos de cadeia média e ácidos graxos insaturados, além do estímulo à prática de exercícios9. A realização de qualquer dieta é um desafio, principalmente para pacientes que têm hiperfagia. Para muitos cuidadores não se vê motivo para realizar tratamento, já que o tipo físico do paciente aparenta ser atlético e, no caso de a doença não ser bem compreendida, essas famílias irão abandonar a terapia devido às adversidades para o tratamento, como a distância a percorrer, qualidade e disponibilidade de transporte, etc. A utilização de atividades lúdicas sobre a conscientização dos fatos médicos da doença ajuda a família a compreender e aprender a conviver com essa doença, com maior adesão às orientações dos profissionais de saúde. O papel do pediatra é fundamental na coordenação da equipe multidisciplinar, que pode englobar profissionais da educação e do direito.
CONCLUSÃO
Paciente tem um quadro característico da SBS caracterizada por três critérios maiores, como presença de lipodistrofia que afeta o tronco, membros e face, aparência acromegaloide e elevada concentração sérica de triglicerídeos, além do teste molecular. O tratamento dessa doença deve ser precoce para evitar complicações, não sendo necessário aguardar pelo teste molecular para ser instituído. Sugerimos a utilização de critérios de diagnóstico clínico. É essencial o aconselhamento genético e ações terapêuticas de uma equipe multidisciplinar formada por profissionais de saúde, educação e direito, que devem ser coordenadas pelo pediatra, que é o profissional da saúde que mais tem contato com a família.
REFERÊNCIAS
1. Loureiro JC, Araújo BC, Duarte RH, Brito AM de, Santos MCO, Pena VL da S, et al. Síndrome de Berardinelli-Seip com acometimento familiar em Patos de Minas - MG: um relato de caso. Brazilian J of Tech. 2022 Sep 10;5(3):181–92.
2. Lima JG de, Campos JTA de M, organizadores. Síndrome de Berardinelli-Seip Aspectos genéticos e morfofisiológicos. 1. ed. Natal: EDUFRN; 2020. p. 51-64.
3. Damasceno E de B, Figueiredo JG de, França JMB, Veras JCD, Borges REA, Melo LP de. Experiência de pessoas que vivem com a Síndrome de Berardinelli-Seip no Nordeste brasileiro. Sci Saúde Coletiva. 2018 Feb;23(2):389–98.
4. Centre de Référence des Pathologies Rares de l’InsulinoSécrétion et de l’Insulino-Sensibilité (PRISIS). Lipodystrophies généralisées congénitales. Paris: Haute Autorité de Santé; 2022 Nov 15.
5. Giori SF, Palhares LGA, Castro IAF, Salgado P de O. Lipodistrofia Generalizada Congênita: Relato de caso. Braz Journal of Surg Clin Research BJSCR. 2019;29(3):29-32.
6. Czepielewski MA, Tavarone V, Riera N, Boschi A, Paula LPD. Síndrome de Berardinelli-Seip: Menino com Baixo Peso e Tecido Subcutâneo Ausente. Clin Biomed Rese. 2010 Nov 19;30(3).
7. Barra C, Savoldelli RD, Manna TD, Kim CA, Magré J, Porta G, et al. Síndrome de Berardinelli-Seip: descrição genética e metabólica de cinco pacientes. Arq Bras Endocrinol Metab. 2011 Feb 1;55(1):54–9.
8. Santos SC dos, Melo US, Lopes SS dos S, Weller M, Kok F. A endogamia explicaria a elevada prevalência de deficiências em populações do Nordeste brasileiro? Sci Saúde Coletiva. 2013 Apr;18(4):1141–50.
9. Bispo MB, Freitas ACC de. Lipodistrofia generalizada congênita: uma revisão bibliográfica. Rese Soc and Develop. 2023 Feb 7;12(2):e21812240288.
1. Centro Universitário Christus (Unichristus) - Fortaleza - CE - Brasil
2. Hospital Infantil Albert Sabin (HIAS), Serviço de Genética Médica - Fortaleza - CE - Brasil
Endereço para correspondência:
Erlane Marques Ribeiro
Centro Universitário Christus (Unichristus).
Av. Dom Luís, 911, Meireles
Fortaleza, CE, Brasil. CEP: 60160-230.
E-mail: erlaneribeiro@yahoo.com.br
Data de Submissão: 02/02/2024
Data de Aprovação: 19/03/2024
Recebido em: 02/02/2024
Aceito em: 19/03/2024
