A Síndrome de Peutz-Jeghers (PJS) é uma condição rara, com padrão de herança autossômica dominante, caracterizada pela presença de pólipos hamartomatosos no trato gastrointestinal, em especial no intestino delgado e com exceção do esôfago, associados à pigmentação mucocutânea característica1,2. A primeira descrição clínica da síndrome remonta a 1921, quando Peutz identificou os primeiros casos, mais tarde aprofundada por Jeghers em 19491.
A prevalência da PJS é estimada entre 1 em 50.000 e 1 em 280.000 nascidos vivos2. A mutação responsável pela síndrome ocorre no gene STK11, localizado no cromossomo 19p13.3, que codifica uma serina/treonina quinase e atua como um supressor tumoral2,3. A perda de heterozigosidade desse gene, observada em pólipos e tumores associados, reforça seu papel na predisposição a neoplasias3.
Os pólipos hamartomatosos, também conhecidos como "pólipos juvenis", são, macroscopicamente, pedunculados, podendo ocorrer esporadicamente ou como parte de síndromes de polipose. A PJS pode apresentar-se com sangramento gastrointestinal, anemia e dor abdominal decorrentes de complicações como intussuscepção, obstrução ou infarto1,3,4.
As manifestações cutâneas são características da síndrome e podem surgir ainda na infância, geralmente antes dos 5 anos, persistindo até a idade adulta1,3. São máculas marrom-escuro ou marrom frequentemente encontradas nos lábios, mas também podem ser observadas na mucosa bucal, pontas dos dedos, falanges e calcanhares. Histopatologicamente, há um aumento no pigmento melanina e nos melanócitos na camada basal da epiderme. Sugere-se que resultem da inflamação que inibe a migração da melanina dos melanócitos para os queratinócitos1.
Apesar do avanço nas pesquisas sobre a PJS, o manejo de complicações, como intussuscepção e o potencial risco aumentado de cânceres associados, ainda representa um desafio clínico significativo. O tratamento cirúrgico, muitas vezes necessário devido à magnitude dos pólipos, continua sendo a principal abordagem terapêutica1,5.
OBJETIVO
O objetivo deste estudo é relatar um caso de um paciente pediátrico com diagnóstico clínico de PJS, quem apresentou um pólipo duodenal gigante e que evoluiu com intussuscepção jejuno-jejunal, ilustrando a complexidade do manejo dessa condição.
MÉTODOS
O estudo baseou-se na revisão do prontuário do paciente e foi aprovado pelo comitê de ética em pesquisa em seres humanos em 2024 (CAAE n. 81479424.3.0000.5264).
RELATO DE CASO
Paciente masculino, de sete anos, atendido no ambulatório de gastroenterologia pediátrica em novembro de 2023, com história de dor abdominal intensa e constipação. Negava sangramento, contudo apresentava episódios de anemia que melhoravam com a reposição de ferro. Familiares próximos, incluindo a mãe, o avô e tios, relatavam histórico de polipose e câncer colorretal. A mãe do paciente aguarda realização de colectomia, e o tio foi submetido a cirurgias por causa de invaginação intestinal. Exames prévios, incluindo ultrassonografia abdominal e exame parasitológico, não revelaram alterações.
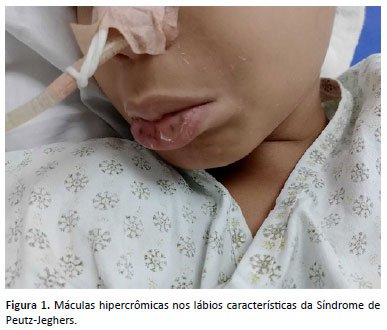
Ao exame físico, o paciente apresentava-se em bom estado geral, eutrófico, com máculas hipercrômicas características da síndrome de Peutz-Jeghers no lábio inferior e na mucosa jugal (Figura 1). A palpação abdominal demonstrou a presença de fezes endurecidas no quadrante inferior esquerdo, sem outras massas palpáveis.
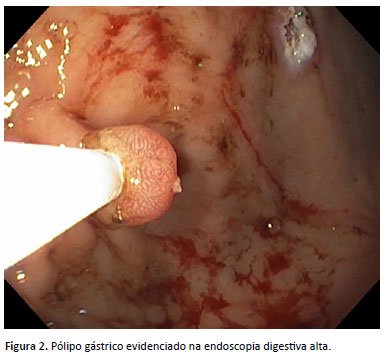
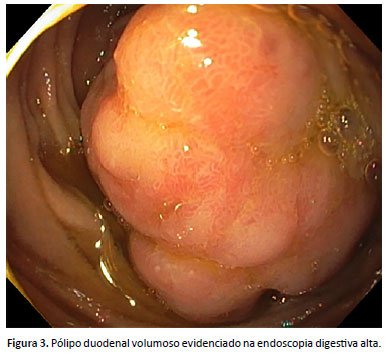
Devido à história familiar e quadro clínico, foi realizada colonoscopia, que evidenciou pólipos sésseis colônicos, classificados como Paris 0-is. O exame histopatológico mostrou que se tratava de pólipos hiperplásicos com alterações inflamatórias e ulcerações. Na consulta subsequente, foi relatada melhora da dor abdominal. Quatro meses após, foi realizada endoscopia digestiva alta que mostrou pólipos sésseis gástricos (Figura 2) e um pólipo séssil duodenal volumoso (Figura 3), ambos classificados como Paris 0-is, sendo realizada polipectomia dos pólipos gástricos e biópsia do pólipo duodenal. O exame histopatológico foi compatível com pólipos hamartomatosos associados à síndrome de Peutz-Jeghers.
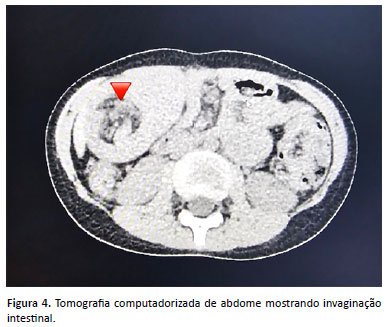
Dois meses após o procedimento, apresentou dor abdominal periumbilical de forte intensidade, motivo pelo qual procurou atendimento médico. Realizou tomografia computadorizada abdominal que revelou invaginação intestinal jejuno-jejunal (Figura 4). Necessitou de intervenção cirúrgica com redução manual, com presença de pólipo como cabeça de invaginação, localizado aproximadamente a 45 cm do ângulo de Treitz. Também foi identificado pólipo ocupando parte da luz a 10 e 90 cm do ângulo de Treitz. Imediatamente foi realizada enterotomia com polipectomia na cabeça da invaginação. Foi feita também enterectomia segmentar com enteroanastomose término-terminal.
O resultado histopatológico do pólipo do jejuno evidenciou processo inflamatório agudo e crônico, com edema parietal, focos de extravasamento hemático e adenocarcinoma em pólipo túbulo-viloso. Após a cirurgia, o paciente evoluiu com queixas ocasionais de dor abdominal e constipação associada.
O paciente está agendado para acompanhamento genético e novas avaliações endoscópicas para monitorar a presença de pólipos e vigilância quanto à evolução.
DISCUSSÃO
A Síndrome de Peutz-Jeghers (SPJ) é um distúrbio hereditário raro caracterizado por pólipos hamartomatosos gastrointestinais6 que demanda uma abordagem abrangente, especialmente em pacientes pediátricos, como o caso do nosso paciente.
Está amplamente relatado que os pólipos de Peutz-Jeghers podem causar sintomas como dor abdominal e vômitos1, o que ficou em concordância com as queixas iniciais do paciente, incluindo dor abdominal intensa e constipação. Outra apresentação comum é o sangramento intestinal, resultante da autoamputação do pólipo, o que ocasiona anemia e, em situações mais graves, múltiplas transfusões sanguíneas6. O paciente não apresentou relato de sangramento, mas episódios de anemia.
A presença de máculas hipercrômicas na mucosa oral e nos lábios é típica da SPJ1,2. A identificação precoce dessas manifestações, aliada ao histórico familiar, facilita o diagnóstico clínico, que pode ser confirmado por testes genéticos, dada a alta taxa de detecção de mutações patogênicas no gene STK112,3.
A presença de história familiar relevante é um fator de risco importante a ser considerado na avaliação clínica, dado que aproximadamente metade dos casos são hereditários, enquanto os restantes são esporádicos como resultado de mutação espontânea6. A história de intervenções cirúrgicas em familiares próximos devido a complicações associadas à polipose ressalta a importância de uma vigilância rigorosa em pacientes pediátricos.
A confirmação por meio de endoscopia digestiva alta e colonoscopia com biópsia e exame histopatológico é fundamental para o diagnóstico, evidenciando pólipos hamartomatosos. Os pólipos são mais comumente encontrados no jejuno (90%), seguido pelo íleo (80%), cólon/reto (50%) e estômago (25%)6. O caso do paciente ilustra a progressão do manejo endoscópico, desde a identificação de pólipos colônicos até a polipectomia de pólipos gástricos e identificação de um pólipo duodenal volumoso.
Embora os pólipos de Peutz-Jeghers sejam, em sua maioria, não neoplásicos, com a formação de feixes musculares dendríticos e hiperplasia epitelial, eles podem resultar em complicações graves, como obstrução intestinal, sangramento e anemia, com destaque para a intussuscepção, uma complicação frequente em crianças4,5. De qualquer forma, o risco de câncer gastrointestinal e extragastrointestinal é alto6, como observado no resultado anatomopatológico do pólipo do jejuno, que evidenciou adenocarcinoma.
Em relação ao tratamento, os pólipos devem ser ressecados endoscopicamente para evitar complicações associadas ao seu crescimento. A ressecção cirúrgica é necessária para pólipos que não podem ser tratados endoscopicamente1. Pólipos do intestino delgado com tamanho superior a 1,5 a 2 cm (ou menores, se causarem sintomas) devem ser removidos para prevenir a intussuscepção3.
Pacientes com intussuscepção sintomática devem ser encaminhados com urgência para intervenção cirúrgica3, como observado neste paciente. A redução radiológica ou endoscópica da intussuscepção não é indicada em crianças sintomáticas com obstrução intestinal devido a um pólipo de PJS. Durante a laparotomia, é recomendável que os pacientes sejam submetidos, preferencialmente, a uma enteroscopia intraoperatória para a remoção de outros pólipos3. A necessidade de laparotomia exploratória e enterectomia neste caso sublinha a importância de um manejo cirúrgico cuidadoso e a necessidade de monitoramento pós-operatório.
O caso reforça a recomendação de vigilância regular e rastreamento para neoplasias gastrointestinal e extragastrointestinal associadas à PJS6. Embora as diretrizes sugiram início do acompanhamento na adolescência, a presença de sintomas e história familiar pode justificar um início mais precoce (8 anos de idade)3, como neste paciente. O acompanhamento genético agendado é crucial, pois a análise de mutações no gene STK11 pode auxiliar na estratificação de risco e na tomada de decisões sobre intervenções preventivas.
REFERÊNCIAS
1. Yamamoto H, Sakamoto H, Kumagai H, Abe T, Ishiguro S, Uchida K, et al. Clinical guidelines for diagnosis and management of Peutz-Jeghers syndrome in children and adults. Digestion. 2023;104(5):335-47.
2. Günay Aslan P, Çağlayan AO, Bora E, Koç A, Yücel H, Ülgenalp A, et al. Clinical and molecular analysis in patients with Peutz-Jeghers syndrome. Turk J Gastroenterol. 2024;35(5):374-84.
3. Latchford A, Cohen S, Auth M, Scaillon M, Viala J, Daniels R, et al. Management of Peutz-Jeghers syndrome in children and adolescents: A position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019;68(3):442-52.
4. Shussman N, Wexner SD. Colorectal polyps and polyposis syndromes. Gastroenterol Rep (Oxf). 2014;2(1):1-15.
5. Ramos-Gonzalez G, Lugo-Rodriguez V, Camacho-Landron C, Rivera-Pedrogo F. Ileo-ileal intussusception caused by hamartomatous polyp. J Pediatr Surg Case Rep. 2021;71:101918.
6. Pandit N, Neupane D, Deo KB. Peutz-Jeghers syndrome: A case series. Int J Surg Case Rep. 2024;122:110117.
1. Instituto de Puericultura e Pediatria Martagão Gesteira da Universidade Federal do Rio de Janeiro, Serviço de Gastroenterologia
2. Hospital Estadual Adão Pereira Nunes - Duque de Caxias - Rio de Janeiro - Brasil
Endereço para correspondência:
Carlos Enrique Crismatt Rodríguez
Instituto de Puericultura e Pediatria Martagão Gesteira
Rua Bruno Lobo, 50, Cidade Universitária
Rio de Janeiro, RJ, Brasil. CEP: 21941-912
E-mail: carloscrismatt44@hotmail.com
Data de Submissão: 11/12/2024
Data de Aprovação: 22/01/2025
Recebido em: 11/12/2024
Aceito em: 22/01/2025
