INTRODUÇÃO
Cistos de colédoco (CC) são dilatações congênitas da árvore biliar, podendo ser intra ou extra-hepáticas. Os CC são raros e apresentam incidências diferentes entre as populações: aproximadamente 1:13.000 na população asiática e 1:100.000 na população ocidental, com maior risco no sexo feminino em comparação ao masculino1.
A apresentação clínica clássica dos CC inclui a tríade de icterícia, dor abdominal e massa palpável no quadrante abdominal superior, sendo essa manifestação mais comum na população pediátrica1,2. O diagnóstico é frequentemente realizado por ultrassonografia (USG), podendo ser complementado por outros exames radiológicos3. O tratamento definitivo consiste na remoção cirúrgica do cisto, sendo o procedimento escolhido e o momento da intervenção determinados pela apresentação clínica, classificação do cisto e idade do paciente4.
A síndrome de Noonan (SN) é uma condição genética de herança autossômica dominante, caracterizada por comprometimento multissistêmico com alta heterogeneidade e expressão clínica variável. Sua tríade clássica inclui alterações faciais, cardiomiopatia hipertrófica e baixa estatura5. A incidência estimada da SN é de 1:1.000 a 1:2.500 nascidos vivos5,6.
A associação entre CC e a SN foi documentada poucas vezes na literatura. Um dos relatos prévios foi publicado em 1993, por C.D. George, descrevendo um caso confirmado por ultrassonografia em um paciente que, além do cisto de colédoco congênito, apresentava rim ectópico e malformação intestinal7.
Dessa forma, considerando a rara apresentação clínica dos cistos de colédoco congênitos, este estudo tem como objetivo relatar um caso envolvendo um cisto de colédoco congênito, detalhar seu diagnóstico, manifestações clínicas e manejo no período neonatal, e documentar a associação entre CC e SN, uma relação com publicações limitadas na literatura.
RELATO DE CASO
Recém-nascido (RN) do sexo masculino, pré-termo, com idade gestacional (IG) de 34 semanas e 3 dias e peso ao nascimento de 2.430g. Filho de mãe primigesta, com 20 anos, hígida. O pré-natal foi iniciado com IG de 21 semanas. Os testes rápidos maternos para sífilis, HIV e hepatite B foram negativos. Na ultrassonografia (USG) morfológica realizada com IG de 22 semanas e 1 dia, foi identificada uma imagem anecoica no fígado, medindo 31x27x28mm, sugestiva de cis-to de colédoco. Além disso, foram observadas hipoplasia do ventrículo direito e da artéria pulmonar, clinodactilia à direita, osso nasal hipoplásico e polidrâmnio.
Uma nova USG foi realizada com IG de 33 semanas e 6 dias, revelando múltiplos dimorfismos: braquicefalia, baixa implantação das orelhas, malformação cardíaca, derrame pleural à esquerda, distorção na arquitetura do tecido renal, criptorquidia e a mesma imagem anecoica no fígado. O cariótipo fetal confirmou 46, XY.
O RN nasceu por cesariana devido a sofrimento fetal, apresentando bradicardia e apneia ao nascimento. Foram realizadas manobras de reanimação neonatal, incluindo intubação orotraqueal na sala de parto e toracocentese de alívio à esquerda. O Apgar foi 2/4/7, e o RN foi encaminhado à UTI Neonatal após estabilização clínica.
Ao exame físico, observaram-se fácies sindrômica, hipertelorismo ocular e mamário, ectrodactilia, abdome globoso com visceromegalias palpáveis, uma massa palpável a 6cm do rebordo costal direito e esplenomegalia a 3cm do rebordo costal esquerdo.
Após o nascimento, exames de imagem complementares foram realizados. O ecocardiograma mostrou miocardiopatia hipertrófica biventricular importante, comunicação interventricular moderada e canal arterial amplo.
Nas imagens realizadas, a radiografia panorâmica (figura 1) revelou pulmões insuflados com derrame pleural bilateral, cardiomegalia e ausência de ar nas alças intestinais. Na ultrassonografia (USG) de abdômen total (Imagens 1b e 1c), observou-se dilatação das vias biliares intra-hepáticas e uma volumosa imagem anecoica de contornos regulares na topografia do hilo hepático, ocupando o hemiabdome direito e medindo aproximadamente 5,9 x 4,8cm, sugestiva de cisto de colédoco. O fígado apresentava topografia, morfologia, dimensões, textura e ecogenicidade normais, com o sistema porta e as veias hepáticas com arquitetura preservada.
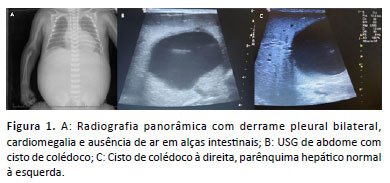
O paciente apresentou dificuldade na aceitação de dieta, não sendo possível afastar compressão extrínseca do trato digestivo pela massa cística. Eliminou mecônio normalmente. Foi avaliado pela genética médica, que levantou a hipótese de síndrome de Noonan, sendo solicitado exame genético para confirmação.
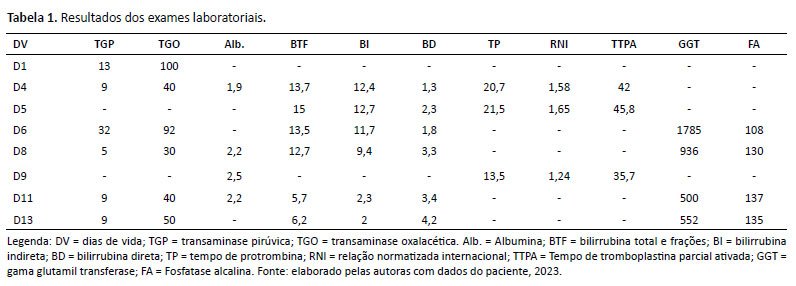
Durante a evolução, o paciente apresentou elevação transitória das enzimas hepáticas, estabilizadas após o quinto dia de vida, mas desenvolveu icterícia colestática progressiva com bilirrubina direta em ascensão, além de alterações no coagulograma (Tabela 1).
Devido às múltiplas alterações e dificuldades na estabilização inicial, o paciente evoluiu com sepse de foco pulmonar, choque séptico refratário e óbito antes de realizar o procedimento cirúrgico para remoção do cisto de colédoco.
O diagnóstico de síndrome de Noonan foi confirmado posteriormente por teste genético, que identificou uma variante patogênica em heterozigose no gene PTPN11.
DISCUSSÃO
Cistos de colédoco (CC) são dilatações congênitas da árvore biliar, podendo ser intra ou extra-hepáticos. Esses cistos são raros e sua etiologia permanece desconhecida, embora existam algumas teorias sobre seu desenvolvimento. Uma das hipóteses sugere uma anormalidade na união dos ductos pancreatobiliares durante o período embrionário, predispondo ao refluxo de suco pancreático para os ductos biliares, o que resulta em inflamação crônica, danos à parede do ducto e alterações císticas8,4. Outras possíveis causas incluem fragilidade da parede do ducto biliar, obstrução distal do ducto biliar, disfunção do esfíncter de Oddi e alterações nas células ganglionares9. A classificação mais amplamente utilizada para os diversos tipos de CC é a classificação de Todani modificada (Tabela 2), de 1977, que categoriza os cistos em cinco tipos (I-V), conforme descrito na literatura10,4.

Clinicamente, a apresentação clássica inclui a tríade de icterícia, dor abdominal e massa palpável no quadrante abdominal superior, frequentemente observada na população pediátrica1,2. Outras manifestações incluem colangite, pancreatite, hipertensão portal, anormalidades na função hepática, coagulopatias e acolia fecal1,2. Em casos raros, pode ocorrer ruptura biliar com peritonite, uma forma de apresentação típica no período neonatal, que exige drenagem de emergência4,9. No caso relatado, o paciente apresentou a tríade clássica logo nos primeiros dias de vida.
O diagnóstico geralmente é realizado por ultrassonografia (USG), podendo ser complementado por tomografia computadorizada (TC), ressonância magnética (RM) ou colangiopancreatografia por ressonância magnética (CPRM), sendo esta última considerada o padrão-ouro para diagnóstico e classificação3,11. No caso do paciente relatado, o diagnóstico foi sugerido no período pré-natal, com imagem anecoica identificada no USG morfológico às 22 semanas de gestação, compatível com cisto de colédoco.
Alterações laboratoriais sugestivas de CC incluem níveis elevados de bilirrubina direta e indireta, TGO, TGP e GGT12. No caso descrito, o paciente apresentou elevação progressiva dos níveis de bilirrubina direta e GGT, mesmo após a primeira semana de vida, enquanto as transaminases mostraram elevações transitórias, possivelmente associadas ao trauma no momento do parto.
O tratamento dos cistos de colédoco é cirúrgico, e o momento ideal para a intervenção é motivo de discussão, especialmente nos casos assintomáticos. Estudos indicam que o diagnóstico precoce, particularmente no período pré-natal, pode influenciar positivamente a escolha do momento da cirurgia e melhorar o prognóstico12. O paciente em questão apresentou sintomas colestáticos, como icterícia, elevação das enzimas hepáticas e compressão extrínseca das vias biliares pelo cisto, indicando a necessidade de intervenção cirúrgica.
A síndrome de Noonan (SN) é uma condição genética autossômica dominante causada por mutações em genes relacionados à sinalização intracelular das proteínas RAS MAPK5. O gene PTPN11 é o mais frequentemente associado à SN6 e foi identificado no caso descrito. A SN apresenta grande variabilidade fenotípica e manifestações multissistêmicas, incluindo alterações faciais características, cardiomiopatia hipertrófica, estenose pulmonar, baixa estatura, criptorquidia, coagulopatias e anomalias renais5,6. Muitos sinais podem ser detectados no período pré-natal, como aumento da translucência nucal, higroma cístico, polidrâmnio, hidropsia fetal e defeitos cardíacos6. Embora o teste genético seja fundamental para o diagnóstico, até 10% dos casos podem apresentar resultados normais, sendo o diagnóstico baseado em alta suspeição clínica13. O paciente relatado apresentou características clássicas da SN, como dismorfias faciais, implantação baixa das orelhas, hipertelorismo, miocardiopatia hipertrófica e criptorquidia. A associação entre SN e cisto de colédoco é extremamente rara, com poucos relatos na literatura, mas é uma característica importante devido ao potencial de degeneração maligna13.
Este relato descreve a associação de duas condições raras, cisto de colédoco congênito e síndrome de Noonan, destacando a importância de uma abordagem multidisciplinar para o manejo de casos complexos. A descrição dessa associação é relevante para aumentar a conscientização na comunidade médica e reforçar a consideração diagnóstica da SN mesmo em casos em que o teste genético seja inconclusivo.
REFERÊNCIAS
1. Soares KC, Arnaoutakis DJ, Kamel I, Rastegar N, Anders R, Maithel S, et al. Choledochal cysts: presentation, clinical differentiation, and management. J Am Coll Surg. 2014;219(6):1167–80. DOI: https://doi.org/10.1016/j. jamcollsurg.2014.04.023.
2. Ye Y, Lui VCH, Tam PKH. Pathogenesis of choledochal cyst: insights from genomics and transcriptomics. Genes (Basel). 2022;13(6):1030. DOI: https://doi.org/10.3390/genes13061030.
3. Murphy AJ, Axt JR, Crapp SJ, Martin CA, Crane GL, Lovvorn HN. Concordance of imaging modalities and cost minimization in the diagnosis of pediatric choledochal cysts. Pediatr Surg Int. 2012;28(6):615–21. DOI: https://doi.org/10.1007/s00383-012-3089-3.
4. Soares KC, Goldstein SD, Ghaseb MA, Kamel I, Hackam DJ, Pawlik TM. Pediatric choledochal cysts: diagnosis and current management. Pediatr Surg Int. 2017;33(6):637–50. DOI: https://doi.org/10.1007/s00383-0174083-6.
5. Carcavilla A, Suarez-Ortega L, Rodriguez Sanchez A, Gonzalez-Casado I, Ramon-Krauel M, Labarta JI, et al. Síndrome de Noonan: actualización genética, clínica y opciones terapéuticas. An Pediatr (Barc). 2020;93(1):61. e1–61.e14. DOI: https://doi.org/10.1016/j.anpedi.2020.04.008.
6. Stuurman KE, Joosten M, van der Burgt I, Elting M, Yntema HG, Meijers-Heijboer H, et al. Prenatal ultrasound findings of rasopathies in a cohort of 424 fetuses: update on genetic testing in the NGS era. J Med Genet. 2019;56(10):654–61. DOI: https://doi.org/10.1136/jmedgenet-2018-105746.
7. George CD, Patton MA, El Sawi M, Sharland M, Adam EJ. Abdominal ultrasound in Noonan syndrome: a study of 44 patients. Pediatr Radiol. 1993;23(4):316–8. DOI: https://doi.org/10.1007/BF02010926.
8. Yamashiro Y, Miyano T, Shimomura H, Suda K, Matsumoto M, Nittono H. Experimental study of the pathogenesis of choledochal cyst and pancreatitis, with special reference to the role of bile acids and pancreatic enzymes in the anomalous choledocho-pancreatic ductal junction. J Pediatr Gastroenterol Nutr. 1984;3(5):721–7. DOI: https://doi.org/10.1002/j.15364801.1984.tb08758.x.
9. Martin RF. Biliary cysts. Surg Clin North Am. 2014;94(2):219–32. DOI: https://doi.org/10.1016/j.suc.2014.01.011.
10. Todani T, Watanabe Y, Narusue M, Tabuchi K, Okajima K. Congenital bile duct cysts. Am J Surg. 1977;134(2):263–9. DOI: https://doi. org/10.1016/0002-9610(77)90359-2.
11. Lewis VA, Adam SZ, Nikolaidis P, Wood C, Wu JG, Yaghmai V, et al. Imaging of choledochal cysts. Abdom Imaging. 2015;40(6):1567–80. DOI: https://doi.org/10.1007/s00261-015-0381-4.
12. Yang D, Li L, Diao M, Xie X, Ming A, Gao R, et al. Risk factors for clinical symptoms of prenatally diagnosed choledochal cysts: a retrospective study. BMC Surg. 2023;23(1):211. DOI: https://doi.org/10.1186/s12893-023-02115-2.
13. Baldo F, Fachin A, Da Re B, Rubinato E, Bobbo M, Barbi E. New insights on Noonan syndrome clinical phenotype: a single-center retrospective study. BMC Pediatr. 2022;22(1):738. DOI: https://doi.org/10.1186/s12887-022-03804-2.
Recebido em: 28/01/2025
Aceito em: 28/01/2025
