A Doença Mista do Tecido Conjuntivo (DMTC) é uma doença autoimune sistêmica com altos títulos de anticorpos anti-U1 ribonucleoproteína (RNP)1-3 associados a manifestações clínicas comuns a mais de uma colagenose, incluindo lúpus eritematoso sistêmico (LES), polimiosite (PM), esclerose sistêmica (ES) e dermatomiosite (DM)4-7. A coexistência dessas afecções em um mesmo indivíduo é raramente relatada, principalmente em crianças. Na maioria dos casos de DMTC, encontramos o LES fazendo parte de seu espectro8.
O objetivo deste artigo é relatar o caso de uma criança, sexo masculino, com DMTC diagnosticada devido a sinais e sintomas compatíveis com esclerose sistêmica e lúpus eritematoso sistêmico.
RELATO DE CASO
D.S.V., sexo masculino, 13 anos, com diagnóstico prévio de esclerose sistêmica com aproximadamente 11,5 anos de idade devido à presença de esclerose cutânea na parte distal dos membros superiores e inferiores, fenômeno de Raynaud, úlceras digitais, microstomia, artrite de joelhos, hipertensão arterial. Fez uso de corticoterapia oral e captopril durante um ano em outro estado, com término em outubro de 2010, e em seguida interrompido tratamento por melhora clínica.
Em julho de 2011, foi internado com quadro de febre, pneumonia com derrame pleural, artrite de joelhos e interfalangeanas proximais, vasculite cutânea (Figura 1) e fenômeno de Raynaud importante (Figura 2). Nesse momento, encontrava-se há 6 meses sem uso de medicações ou acompanhamento médico.
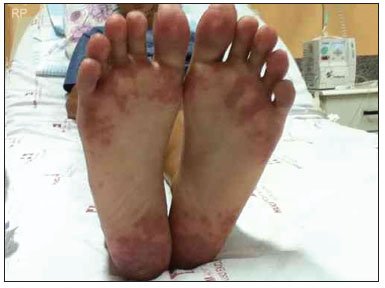
Figura 1. Vasculite cutânea.
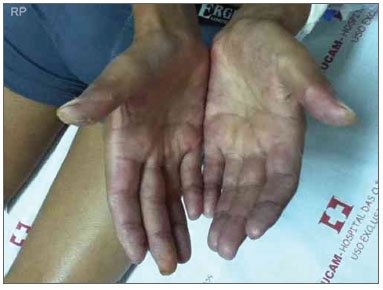
Figura 2. Fenômeno de Raynaud.
Evoluiu rapidamente para piora clínica, com poliserosite (derrame pleural, pericárdico e ascite), hipertensão arterial, diarreia, convulsão, lesão renal aguda multifatorial não dialítica com recuperação total da função renal e não caracterizada crise renal esclerodérmica, insuficiência cardíaca, doença do refluxo gastroesofágico.
Exames apontavam anticorpo antinuclear (FAN) 1:640 padrão nuclear pontilhado grosso,anti-Smith (anti-Sm) 1:400(valorde referência < 1:50), anti-RNP 1:409.600 (valor de referência < 1:50), anticardiolipina IgM 16,1 MPL (indeterminado: 12,5-19)/IgG 14,6 GPL (negativo: < 15), anemia hemolítica, linfopenia, proteinúria não nefrótica 740 mg/24h, tomografia (TC) de tórax de alta resolução com fibrose pulmonar, TC de crânio com vasculite, espirometria com padrão obstrutivo grave, ecocardiograma com processo agudo pancardíaco - pericardite, endocardite e miocardite, sem descrição de hipertensão pulmonar.
Fez uso de captopril, enalapril, losartan, cedilanide, omeprazol, bromoprida, metilprednisolona (2mg/kg/dia) 17 dias, oxacilina 9 dias, B 11 dias.
Paciente foi gradativamente piorando, mesmo em uso das medicações acima. Ficou internado por 48 dias, sendo 36 em UTI pediátrica, necessitando de ventilação mecânica por 14 dias, uso de drogas vasoativas, drenagem de tórax e politransfusão de concentrado de hemácias.
Óbito em 31 de agosto de 2011, apresentando como causa mortis sepse.
DISCUSSÃO
A DMTC apresenta etiologia desconhecida9, é rara entre crianças8,10 e, segundo o banco de dados da reumatologia pediátrica dos EUA, constitui 0,3%-0,6% dos pacientes reumatológicos, dados estes não disponíveis no Brasil4,5,10.
De acordo com a investigação diagnóstica, o quadro apresentado sugere DMTC, composta por altos títulos de anti-RNP e sinais e sintomas de esclerose sistêmica (esclerose cutânea distal dos membros, fenômeno de Raynaud, úlceras digitais, insuficiência cardíaca, artrite, hipertensão, fibrose pulmonar11, FAN positivo, DRGE) e LES (poliartrite, serosite, anemia hemolítica, linfopenia, convulsão, proteinúria, FAN e anti-SM positivos).
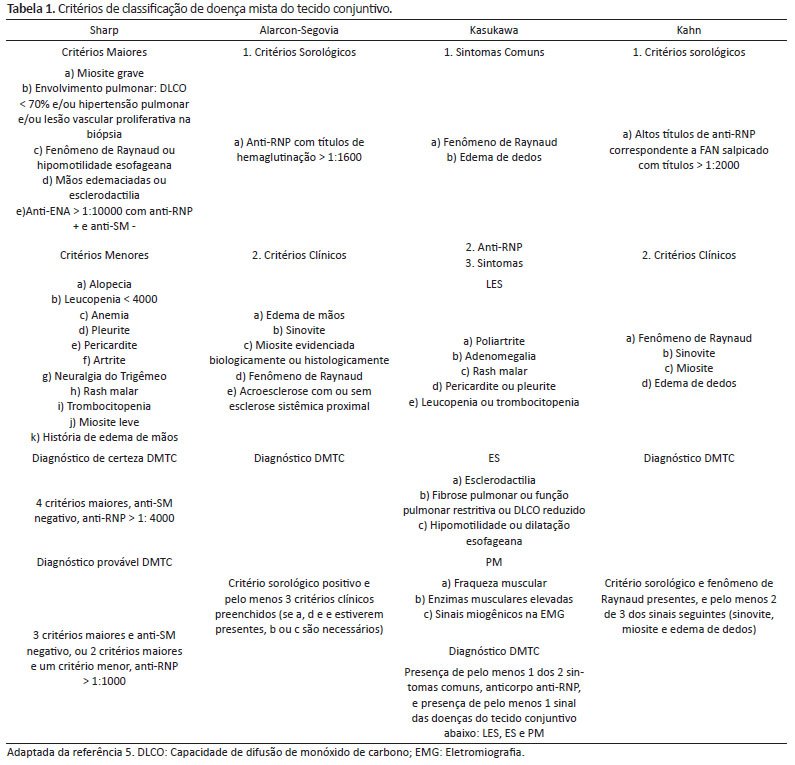
A definição de DMTC não é padronizada universalmente4,12, visto que existem vários critérios de classificação para a doença (Tabela 1). Apenas o critério de Sharp considera que o autoanticorpo anti-SM deve ser obrigatoriamente negativo, o que invalidaria nosso diagnóstico final. Portanto, considerando a classificação de Alarcon-Segovia e de Kasukawa, que são as mais utilizadas em pediatria4,5,10,13, classificamos o paciente como portador de DMTC.
A DMTC é mais comum entre as mulheres do que nos homens5,8,14. A idade média de início da doença costuma ser de 10,7 anos (variando de 6,5 a 14 anos)10. Nenhuma distribuição étnica é conhecida4.
A apresentação clínica da DMTC na infância pode variar de uma doença aguda grave imitando uma sepse, a uma doença de início insidioso com acumulação gradual de sinais e sintomas durante meses a anos. Deve-se suspeitar de DMTC em crianças com doença multissistêmica sem causa aparente8.
Não há nenhum tratamento específico para a DMTC5,6,10 e a medicação destina-se aos sintomas específicos de cada colagenose envolvida4. Muitos pacientes recebem bloqueadores dos canais de cálcio para fenômeno de Raynaud. A maioria das crianças responde com baixas doses de esteroides, anti-inflamatórios não esteroidais e hidroxicloroquina4,5,8,15. Uma combinação destes e de vários outros imunossupressores é usada quando há envolvimento de diferentes órgãos, incluindo o metotrexato, micofenolato mofetil, azatioprina, ciclofosfamida, ciclosporina e agentes biológicos4,6.
CONCLUSÃO
DMTC é uma doença rara na infância e, principalmente, no sexo masculino. O caso descrito compreende um quadro grave de associação de sinais e sintomas de duas colagenoses, a saber: lúpus eritematoso sistêmico e esclerose sistêmica. Como havia título alto de anti-RNP, o diagnóstico foi dado como DMTC. Apesar do uso de corticoterapia venosa, antibióticos de amplo espectro, vasodilatadores e anti-hipertensivos, o paciente evoluiu com piora progressiva e óbito. Os pediatras devem suspeitar de DMTC em crianças com doença multissistêmica sem causa aparente, visto que o diagnóstico precoce e o tratamento são fundamentais na redução da morbimortalidade da doença.
REFERÊNCIAS
1. Hoffman RW, Maldonado ME. Immune pathogenesis of Mixed Connective Tissue Disease: a short analytical review. Clin Immunol. 2008;128(1):8-17. DOI: http://dx.doi.org/10.1016/j.clim.2008.03.461
2. Hoffman RW, Greidinger EL. Mixed connective tissue disease. Curr Opin Rheumatol. 2000;12(5):386-90. DOI: http://dx.doi.org/10.1097/00002281-200009000-00006
3. Aringer M, Steiner G, Smolen JS. Does mixed connective tissue disease exist? Yes. Rheum Dis Clin North Am. 2005;31(3):411-20. DOI: http://dx.doi.org/10.1016/j.rdc.2005.04.007
4. Swart JF, Wulffraat NM. Diagnostic workup for mixed connective tissue disease in childhood. Isr Med Assoc J. 2008;10(8-9):650-2.
5. Pepmueller PH, Lindsley CB, Cassidy JT. Mixed connective tissue disease and undifferentiated connective tissue disease. In: Cassidy JT, Laxer RM, Petty RE, Lindsey CB, eds. Cassidy: Textbook of Pediatric Rheumatology. 6th ed. Philadelphia: Saunders; 2011. p.448-57.
6. Gensch K, Gudowius S, Niehues T, Kuhn A. Connective tissue diseases in childhood. Hautarzt. 2005;56(10):925-36. PMID: 16160808 DOI: http://dx.doi.org/10.1007/s00105-005-1016-4
7. Cappelli S, Bellando Randone S, Martinović D, Tamas MM, Pasalić K, Allanore Y, et al. "To be or not to be," ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum. 2012;41(4):589-98. DOI: http://dx.doi.org/10.1016/j.semar-thrit.2011.07.010
8. Kumar TS, Aggarwal A. Approach to a patient with connective tissue disease. Indian J Pediatr. 2010;77(10):1157-64. PMID: 20924723 DOI: http://dx.doi.org/10.1007/s12098-010-0207-x
9. Gunnarsson R, Molberg O, Gilboe IM, Gran JT; PAHNOR1 Study Group. The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis. 2011;70(6):1047-51. DOI: http://dx.doi.org/10.1136/ard.2010.143792
10. Tsai YY, Yang YH, Yu HH, Wang LC, Lee JH, Chiang BL. Fifteen-year experience of pediatric-onset mixed connective tissue disease. Clin Rheumatol. 2010;29(1):53-8. DOI: http://dx.doi.org/10.1007/s10067-009-1276-y
11. García-Peña P, Boixadera H, Barber I, Toran N, Lucaya J, Enríquez G. Thoracic findings of systemic diseases at high-resolution CT in children. Radiographics. 2011;31(2):465-82. DOI: http://dx.doi.org/10.1148/rg.312095160
12. Maldonado ME, Perez M, Pignac-Kobinger J, Marx ET, Tozman EM, Greidinger EL, et al. Clinical and immunologic manifestations of mixed connective tissue disease in a Miami population compared to a Midwestern US Caucasian population. J Rheumatol. 2008;35(3):429-37.
13. Amigues JM, Cantagrel A, Abbal M, Mazieres B. Comparative study of 4 diagnosis criteria sets for mixed connective tissue disease in patients with anti-RNP antibodies. Autoimmunity Group of the Hospitals of Toulouse. J Rheumatol. 1996;23(12):2055-62.
14. Gaubitz M. Epidemiology of connective tissue disorders. Rheumatology (Oxford). 2006;45 Suppl 3:iii3-4. PMID: 16987829 DOI: http://dx.doi.org/10.1093/rheumatology/kel282
15. Vaessen S, Hoyoux C, Kaye O, Lepage P. Mixed connective tissue disease in childhood: report of two clinical cases. Rev Med Liege. 2004;59(11):648-52.
1. Pediatra - Residente em Neonatologia. Hospital Universitário Cassiano Antonio de Moraes. Universidade Federal do Espírito Santo
2. Nefrologista Pediátrica do Hospital Universitário Cassiano Antônio de Moraes/Universidade Federal do Espírito Santo (HUCAM/UFES)
3. Reumatologista Pediátrica - Reumatologista Pediátrica do Hospital Estadual Infantil Nossa Senhora da Glória (HEINSG)
Endereço para correspondência:
Rafaela Iglesias
Hospital Universitário Cassiano Antônio de Moraes/Universidade Federal do Espírito Santo (HUCAM/UFES)
Rua Hamilton Almeida Guimarães, nº 245, Morada de Camburi
Vitória - ES. Brasil. CEP: 29062-525
